

Deep RNA sequencing of L. monocytogenes reveals overlapping and extensive stationary phase and sigma B-dependent transcriptomes, including multiple highly transcribed noncoding RNAs
- PMID: 20042087
- PMCID: PMC2813243
- DOI: 10.1186/1471-2164-10-641
Deep RNA sequencing of L. monocytogenes reveals overlapping and extensive stationary phase and sigma B-dependent transcriptomes, including multiple highly transcribed noncoding RNAs
Abstract
Background: Identification of specific genes and gene expression patterns important for bacterial survival, transmission and pathogenesis is critically needed to enable development of more effective pathogen control strategies. The stationary phase stress response transcriptome, including many sigmaB-dependent genes, was defined for the human bacterial pathogen Listeria monocytogenes using RNA sequencing (RNA-Seq) with the Illumina Genome Analyzer. Specifically, bacterial transcriptomes were compared between stationary phase cells of L. monocytogenes 10403S and an otherwise isogenic DeltasigB mutant, which does not express the alternative sigma factor sigmaB, a major regulator of genes contributing to stress response, including stresses encountered upon entry into stationary phase.
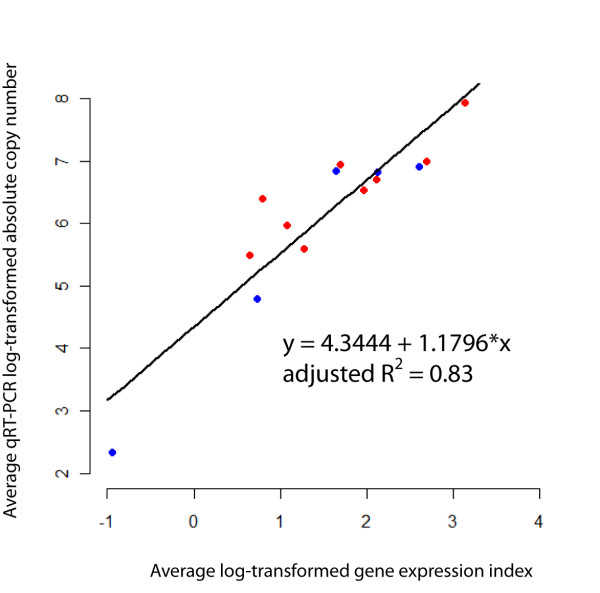
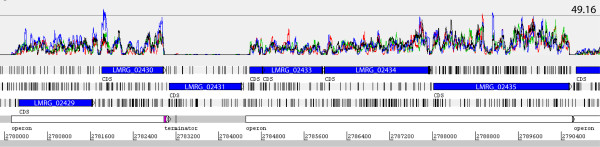
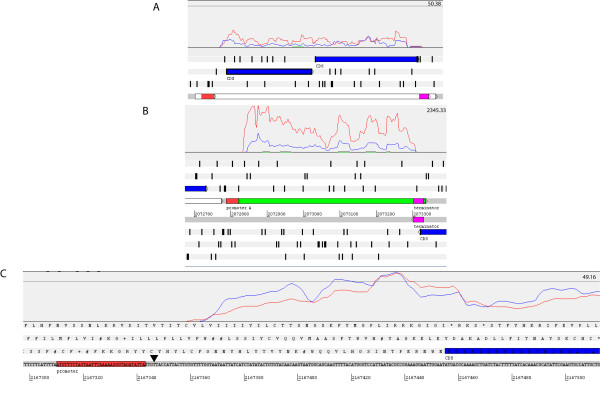
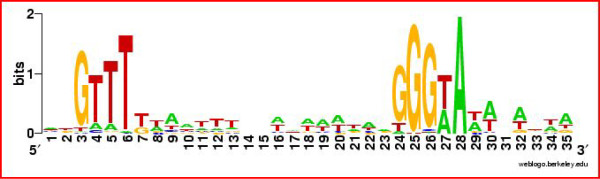
Results: Overall, 83% of all L. monocytogenes genes were transcribed in stationary phase cells; 42% of currently annotated L. monocytogenes genes showed medium to high transcript levels under these conditions. A total of 96 genes had significantly higher transcript levels in 10403S than in DeltasigB, indicating sigmaB-dependent transcription of these genes. RNA-Seq analyses indicate that a total of 67 noncoding RNA molecules (ncRNAs) are transcribed in stationary phase L. monocytogenes, including 7 previously unrecognized putative ncRNAs. Application of a dynamically trained Hidden Markov Model, in combination with RNA-Seq data, identified 65 putative sigmaB promoters upstream of 82 of the 96 sigmaB-dependent genes and upstream of the one sigmaB-dependent ncRNA. The RNA-Seq data also enabled annotation of putative operons as well as visualization of 5'- and 3'-UTR regions.
Conclusions: The results from these studies provide powerful evidence that RNA-Seq data combined with appropriate bioinformatics tools allow quantitative characterization of prokaryotic transcriptomes, thus providing exciting new strategies for exploring transcriptional regulatory networks in bacteria.
Figures


References
-
- Watkins J, Sleath KP. Isolation and enumeration of Listeria monocytogenes from sewage, sewage sludge and river water. J Appl Bacteriol. 1981;50(1):1–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
