

Perspective on Diabatic Models of Chemical Reactivity as Illustrated by the Gas-Phase S(N)2 Reaction of Acetate Ion with 1,2-Dichloroethane
- PMID: 20047005
- PMCID: PMC2658610
- DOI: 10.1021/ct800318h
Perspective on Diabatic Models of Chemical Reactivity as Illustrated by the Gas-Phase S(N)2 Reaction of Acetate Ion with 1,2-Dichloroethane
Abstract
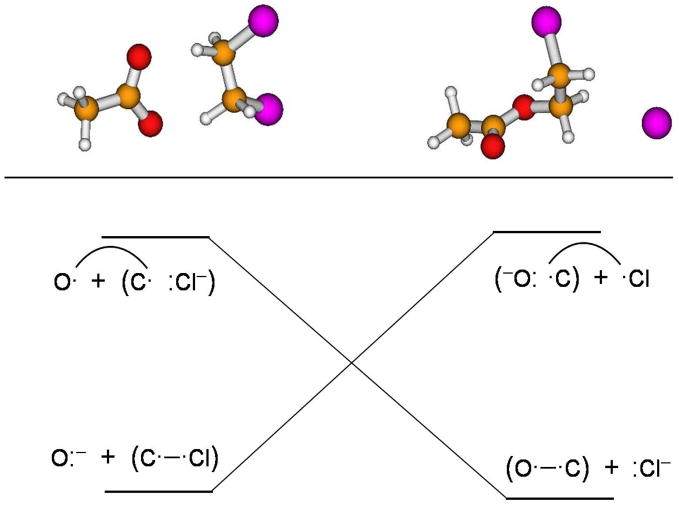
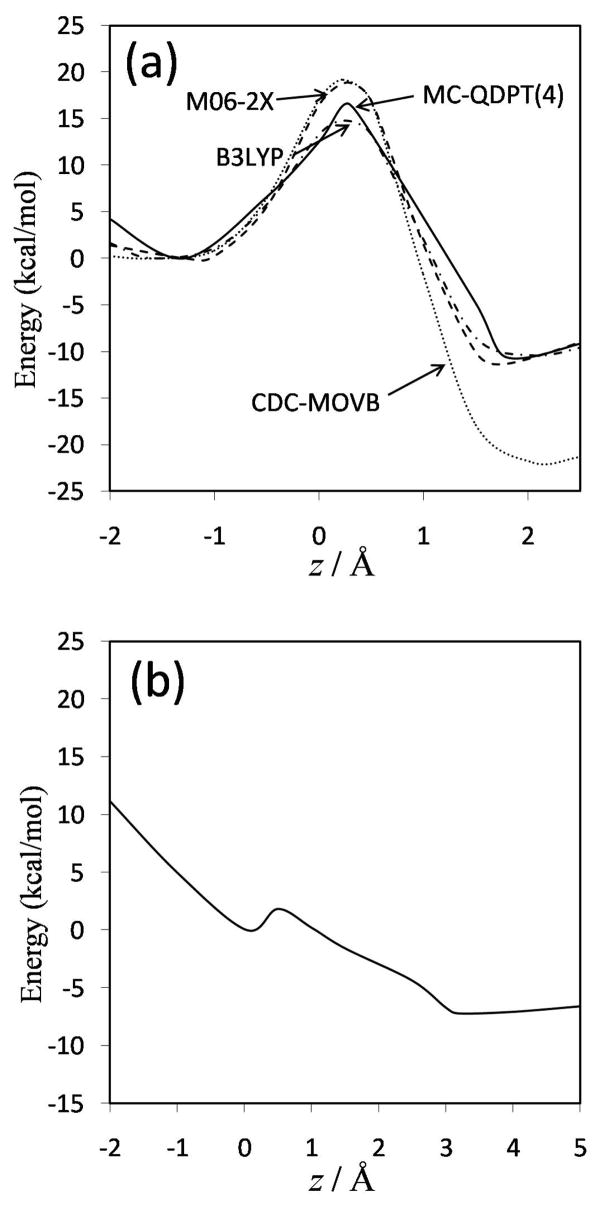
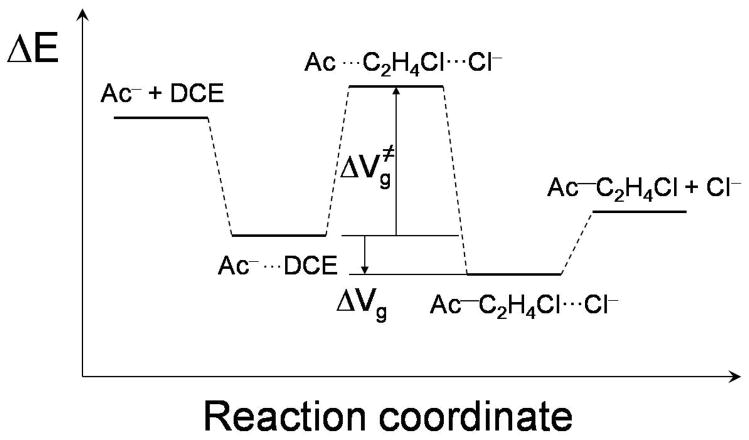
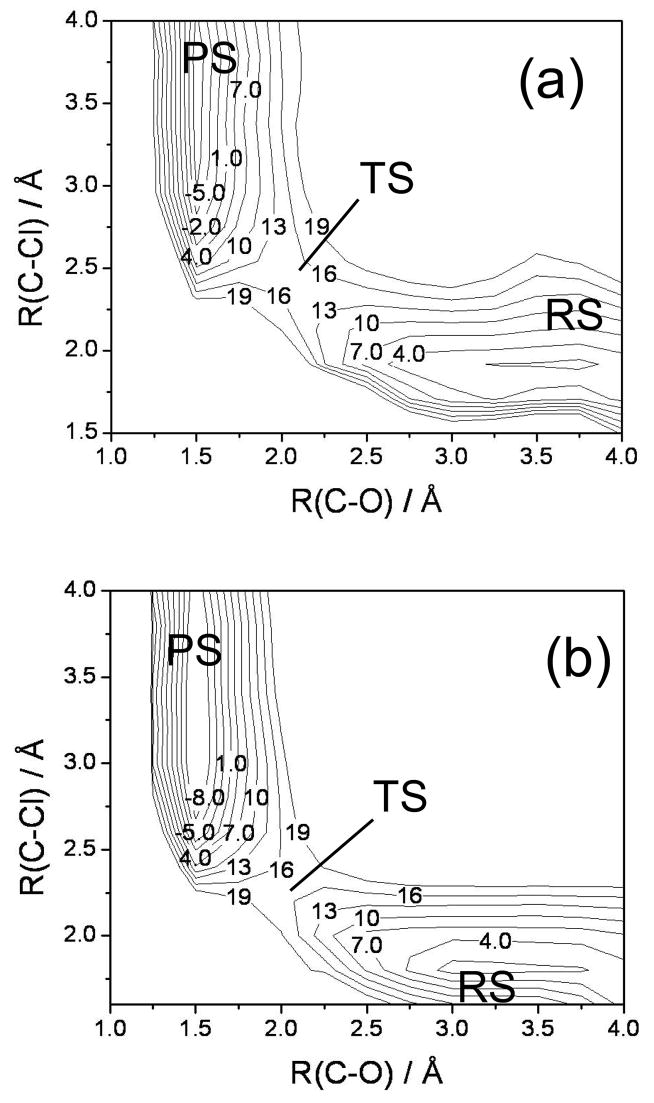
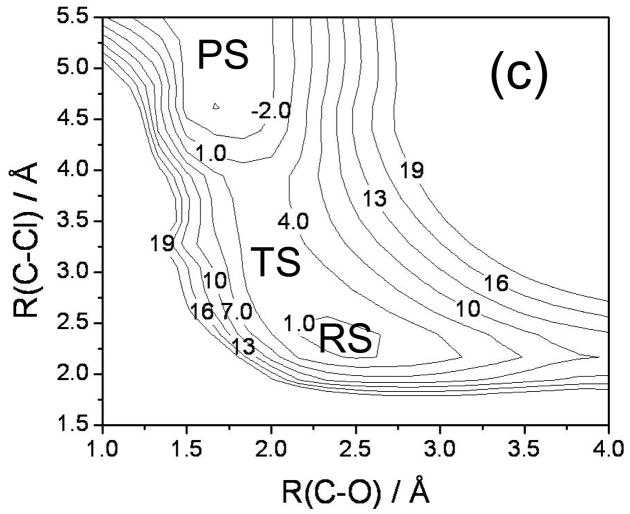
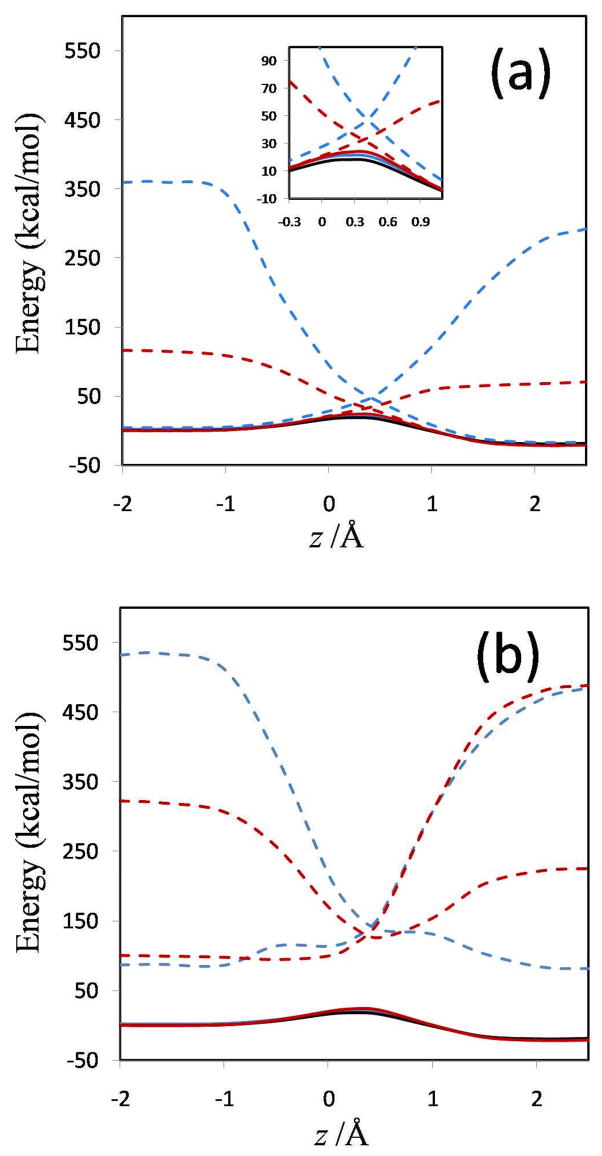
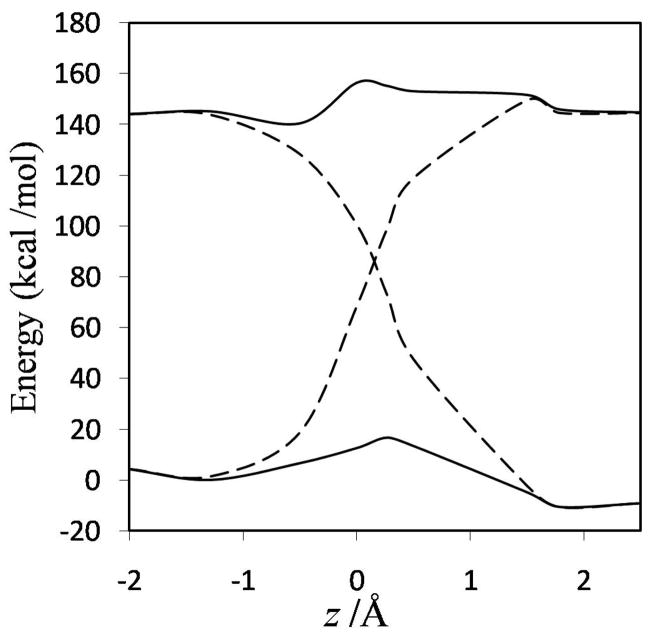
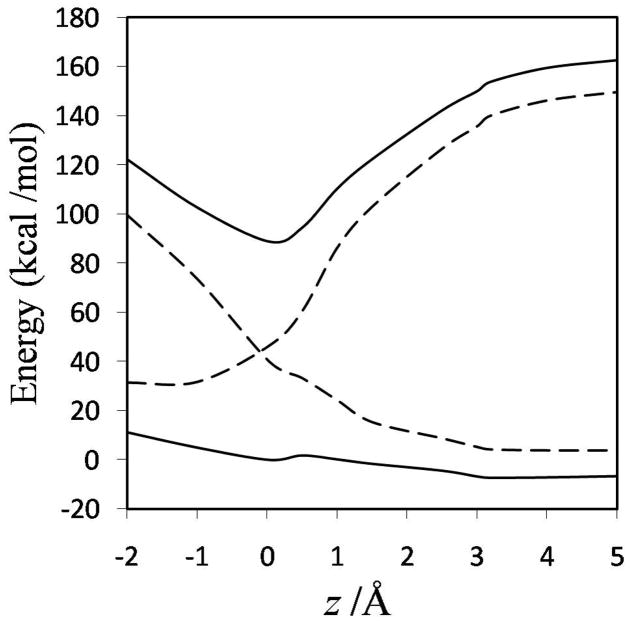
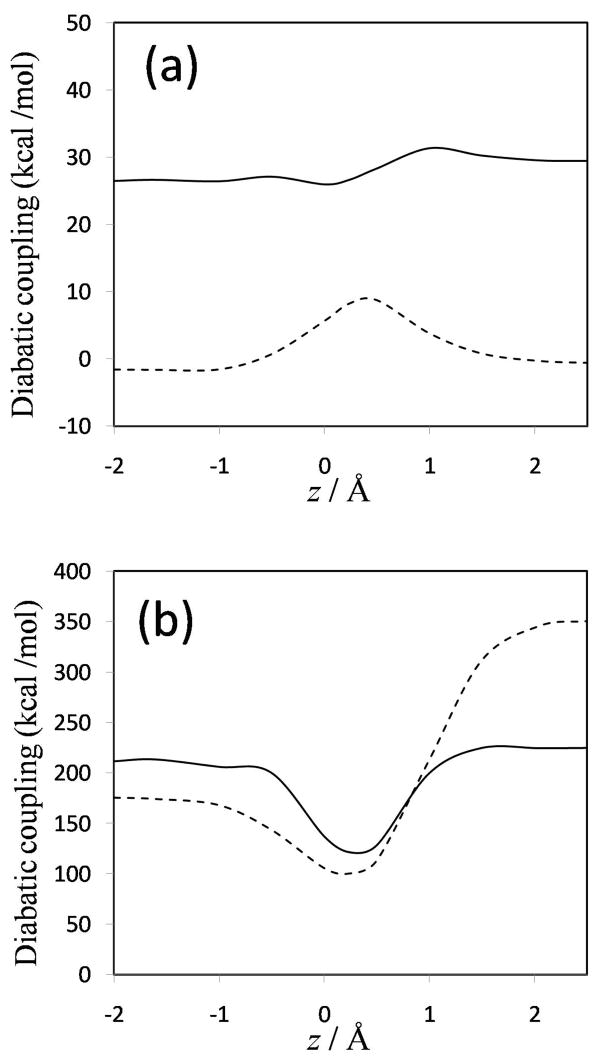
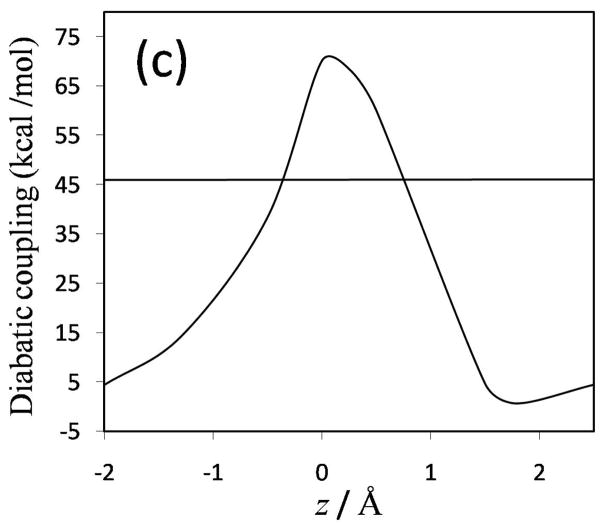
Diabatic models are widely employed for studying chemical reactivity in condensed phases and enzymes, but there has been little discussion of the pros and cons of various diabatic representations for this purpose. Here we discuss and contrast six different schemes for computing diabatic potentials for a charge rearrangement reaction. They include (i) the variational diabatic configurations (VDC) constructed by variationally optimizing individual valence bond structures and (ii) the consistent diabatic configurations (CDC) obtained by variationally optimizing the ground-state adiabatic energy, both in the nonorthogonal molecular orbital valence bond (MOVB) method, along with the orthogonalized (iii) VDC-MOVB and (iv) CDC-MOVB models. In addition, we consider (v) the fourfold way (based on diabatic molecular orbitals and configuration uniformity), and (vi) empirical valence bond (EVB) theory. To make the considerations concrete, we calculate diabatic electronic states and diabatic potential energies along the reaction path that connects the reactant and the product ion-molecule complexes of the gas-phase bimolecular nucleophilic substitution (S(N)2) reaction of 1,2-dichloethane (DCE) with acetate ion, which is a model reaction corresponding to the reaction catalyzed by haloalkane dehalogenase. We utilize ab initio block-localized molecular orbital theory to construct the MOVB diabatic states and ab initio multi-configuration quasidegenerate perturbation theory to construct the fourfold-way diabatic states; the latter are calculated at reaction path geometries obtained with the M06-2X density functional. The EVB diabatic states are computed with parameters taken from the literature. The MOVB and fourfold-way adiabatic and diabatic potential energy profiles along the reaction path are in qualitative but not quantitative agreement with each other. In order to validate that these wave-function-based diabatic states are qualitatively correct, we show that the reaction energy and barrier for the adiabatic ground state, obtained with these methods, agree reasonably well with the results of high-level calculations using the composite G3SX and G3SX(MP3) methods and the BMC-CCSD multi-coefficient correlation method. However, a comparison of the EVB gas-phase adiabatic ground-state reaction path with those obtained from MOVB and with the fourfold way reveals that the EVB reaction path geometries show a systematic shift towards the products region, and that the EVB lowest-energy path has a much lower barrier. The free energies of solvation and activation energy in water reported from dynamical calculations based on EVB also imply a low activation barrier in the gas phase. In addition, calculations of the free energy of solvation using the recently proposed SM8 continuum solvation model with CM4M partial atomic charges lead to an activation barrier in reasonable agreement with experiment only when the geometries and the gas-phase barrier are those obtained from electronic structure calculations, i.e., methods i-v. These comparisons show the danger of basing the diabatic states on molecular mechanics without the explicit calculation of electronic wave functions. Furthermore, comparison of schemes i-v with one another shows that significantly different quantitative results can be obtained by using different methods for extracting diabatic states from wave function calculations, and it is important for each user to justify the choice of diabatization method in the context of its intended use.
Figures
Similar articles
-
On the construction of diabatic and adiabatic potential energy surfaces based on ab initio valence bond theory.J Phys Chem A. 2008 Dec 18;112(50):12925-35. doi: 10.1021/jp803050e. J Phys Chem A. 2008. PMID: 18828577 Free PMC article.
-
An Effective Hamiltonian Molecular Orbital-Valence Bond (MOVB) Approach for Chemical Reactions Applied to the Nucleophilic Substitution Reaction of Hydrosulfide Ion and Chloromethane.J Chem Theory Comput. 2009 Jan 1;5(1):174-185. doi: 10.1021/ct800421y. J Chem Theory Comput. 2009. PMID: 20047006 Free PMC article.
-
A Non-Orthogonal Block-Localized Effective Hamiltonian Approach for Chemical and Enzymatic Reactions.J Chem Theory Comput. 2010 Jul 13;6(7):2242-2251. doi: 10.1021/ct1001686. J Chem Theory Comput. 2010. PMID: 20694172 Free PMC article.
-
A diabatic representation including both valence nonadiabatic interactions and spin-orbit effects for reaction dynamics.J Phys Chem A. 2007 Sep 6;111(35):8536-51. doi: 10.1021/jp072590u. Epub 2007 Aug 11. J Phys Chem A. 2007. PMID: 17691756
-
Diabatic-At-Construction Method for Diabatic and Adiabatic Ground and Excited States Based on Multistate Density Functional Theory.J Chem Theory Comput. 2017 Mar 14;13(3):1176-1187. doi: 10.1021/acs.jctc.6b01176. Epub 2017 Feb 13. J Chem Theory Comput. 2017. PMID: 28135420 Free PMC article.
Cited by
-
The EVB as a quantitative tool for formulating simulations and analyzing biological and chemical reactions.Faraday Discuss. 2010;145:71-106. doi: 10.1039/B907354J. Faraday Discuss. 2010. PMID: 25285029 Free PMC article.
-
Biochemistry and theory of proton-coupled electron transfer.Chem Rev. 2014 Apr 9;114(7):3381-465. doi: 10.1021/cr4006654. Epub 2014 Apr 1. Chem Rev. 2014. PMID: 24684625 Free PMC article. Review. No abstract available.
-
Energy decomposition analysis based on a block-localized wavefunction and multistate density functional theory.Phys Chem Chem Phys. 2011 Apr 21;13(15):6760-75. doi: 10.1039/c0cp02206c. Epub 2011 Mar 2. Phys Chem Chem Phys. 2011. PMID: 21369567 Free PMC article. Review.
-
On unjustifiably misrepresenting the EVB approach while simultaneously adopting it.J Phys Chem B. 2009 Aug 6;113(31):10905-15. doi: 10.1021/jp901709f. J Phys Chem B. 2009. PMID: 19606825 Free PMC article.
-
On the Interfragment Exchange in the X-Pol Method.J Chem Theory Comput. 2010;6(8):2469-2476. doi: 10.1021/ct100268p. J Chem Theory Comput. 2010. PMID: 20730021 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous