

Phylogenetic comparative assembly
- PMID: 20047659
- PMCID: PMC2826331
- DOI: 10.1186/1748-7188-5-3
Phylogenetic comparative assembly
Abstract
Background: Recent high throughput sequencing technologies are capable of generating a huge amount of data for bacterial genome sequencing projects. Although current sequence assemblers successfully merge the overlapping reads, often several contigs remain which cannot be assembled any further. It is still costly and time consuming to close all the gaps in order to acquire the whole genomic sequence.
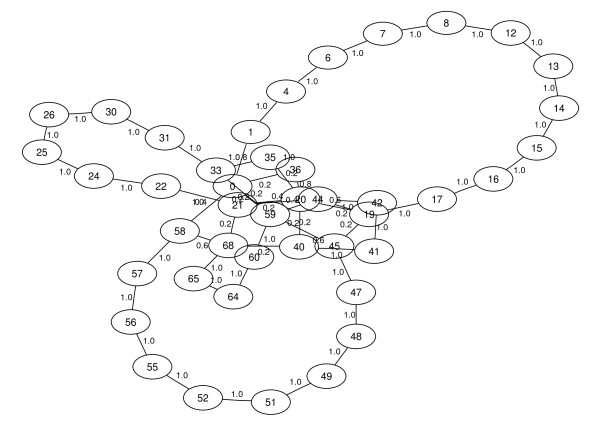
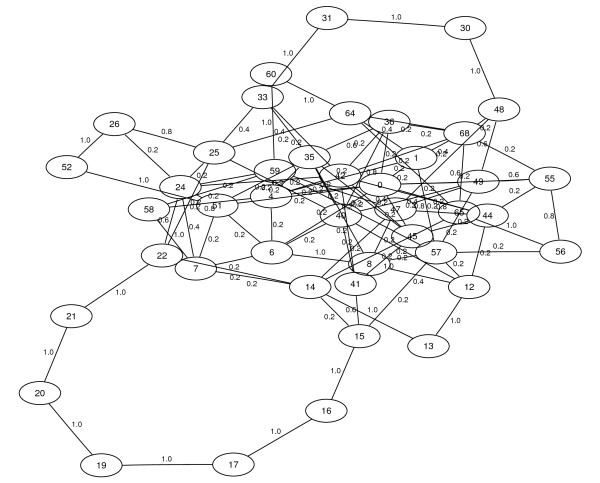
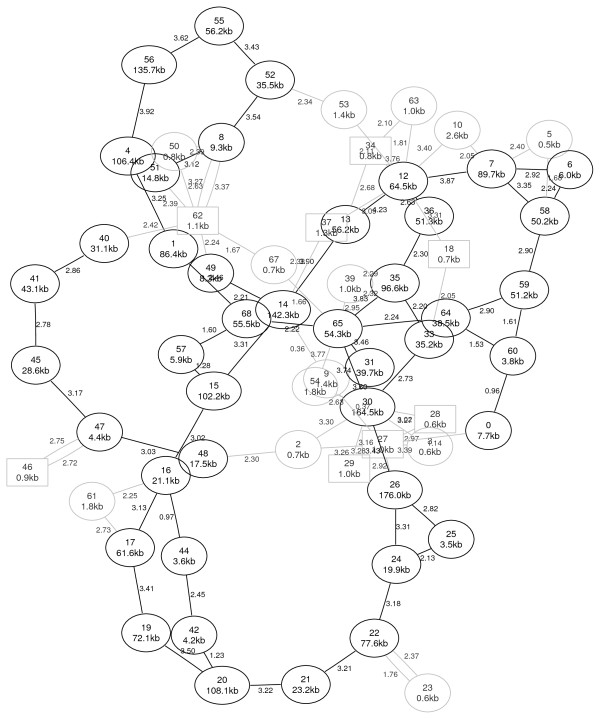
Results: Here we propose an algorithm that takes several related genomes and their phylogenetic relationships into account to create a graph that contains the likelihood for each pair of contigs to be adjacent. Subsequently, this graph can be used to compute a layout graph that shows the most promising contig adjacencies in order to aid biologists in finishing the complete genomic sequence. The layout graph shows unique contig orderings where possible, and the best alternatives where necessary.
Conclusions: Our new algorithm for contig ordering uses sequence similarity as well as phylogenetic information to estimate adjacencies of contigs. An evaluation of our implementation shows that it performs better than recent approaches while being much faster at the same time.
Figures

Similar articles
-
Fragmentation and Coverage Variation in Viral Metagenome Assemblies, and Their Effect in Diversity Calculations.Front Bioeng Biotechnol. 2015 Sep 17;3:141. doi: 10.3389/fbioe.2015.00141. eCollection 2015. Front Bioeng Biotechnol. 2015. PMID: 26442255 Free PMC article.
-
Graph analysis of fragmented long-read bacterial genome assemblies.Bioinformatics. 2019 Nov 1;35(21):4239-4246. doi: 10.1093/bioinformatics/btz219. Bioinformatics. 2019. PMID: 30918948
-
FastEtch: A Fast Sketch-Based Assembler for Genomes.IEEE/ACM Trans Comput Biol Bioinform. 2019 Jul-Aug;16(4):1091-1106. doi: 10.1109/TCBB.2017.2737999. Epub 2017 Sep 11. IEEE/ACM Trans Comput Biol Bioinform. 2019. PMID: 28910776
-
Contig-Layout-Authenticator (CLA): A Combinatorial Approach to Ordering and Scaffolding of Bacterial Contigs for Comparative Genomics and Molecular Epidemiology.PLoS One. 2016 Jun 1;11(6):e0155459. doi: 10.1371/journal.pone.0155459. eCollection 2016. PLoS One. 2016. PMID: 27248146 Free PMC article.
-
METAMVGL: a multi-view graph-based metagenomic contig binning algorithm by integrating assembly and paired-end graphs.BMC Bioinformatics. 2021 Jul 22;22(Suppl 10):378. doi: 10.1186/s12859-021-04284-4. BMC Bioinformatics. 2021. PMID: 34294039 Free PMC article.
Cited by
-
Linearization of ancestral multichromosomal genomes.BMC Bioinformatics. 2012;13 Suppl 19(Suppl 19):S11. doi: 10.1186/1471-2105-13-S19-S11. Epub 2012 Dec 19. BMC Bioinformatics. 2012. PMID: 23281593 Free PMC article.
-
The inference of gene trees with species trees.Syst Biol. 2015 Jan;64(1):e42-62. doi: 10.1093/sysbio/syu048. Epub 2014 Jul 28. Syst Biol. 2015. PMID: 25070970 Free PMC article. Review.
-
r2cat: synteny plots and comparative assembly.Bioinformatics. 2010 Feb 15;26(4):570-1. doi: 10.1093/bioinformatics/btp690. Epub 2009 Dec 16. Bioinformatics. 2010. PMID: 20015948 Free PMC article.
-
Comparative genomics approach to detecting split-coding regions in a low-coverage genome: lessons from the chimaera Callorhinchus milii (Holocephali, Chondrichthyes).Brief Bioinform. 2011 Sep;12(5):474-84. doi: 10.1093/bib/bbr038. Epub 2011 Jun 28. Brief Bioinform. 2011. PMID: 21712341 Free PMC article.
-
Genome reassembly with high-throughput sequencing data.BMC Genomics. 2013;14 Suppl 1(Suppl 1):S8. doi: 10.1186/1471-2164-14-S1-S8. Epub 2013 Jan 21. BMC Genomics. 2013. PMID: 23368744 Free PMC article.
References
-
- Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24(3):133–141. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
