

Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits
- PMID: 20047948
- PMCID: PMC2830832
- DOI: 10.1093/hmg/ddp580
Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits
Abstract
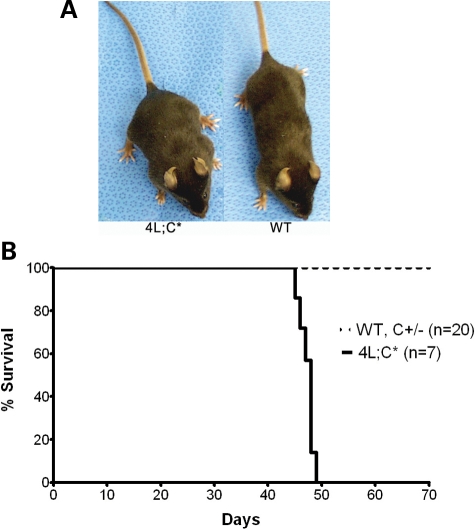
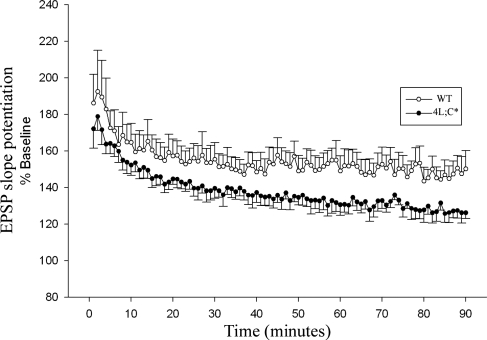
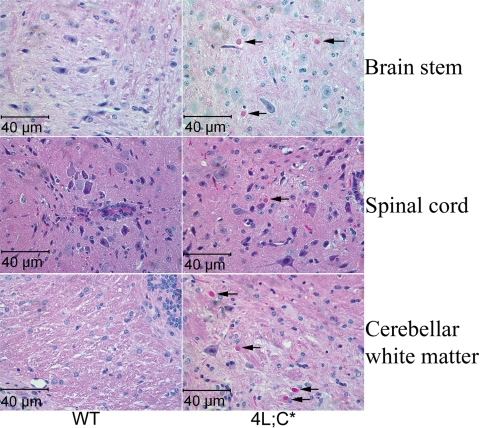
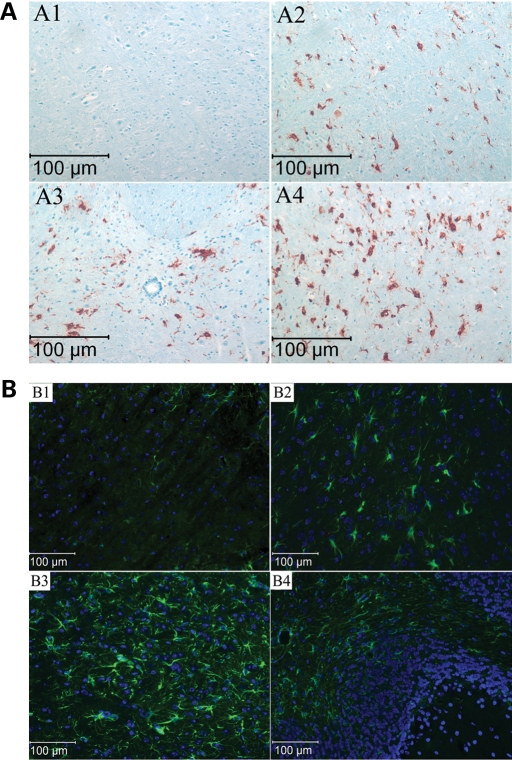
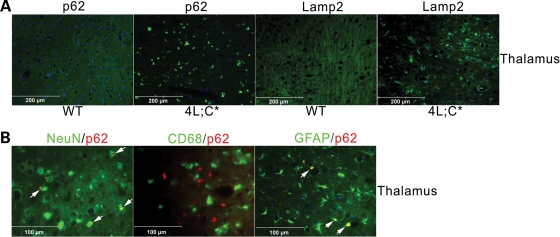
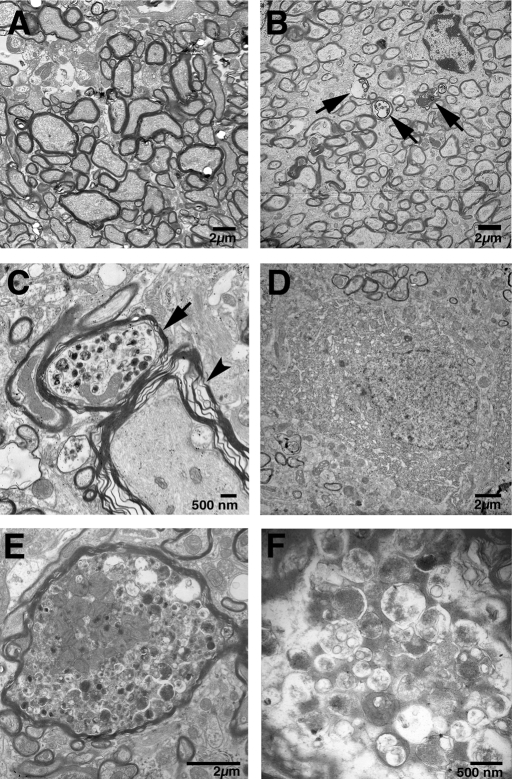
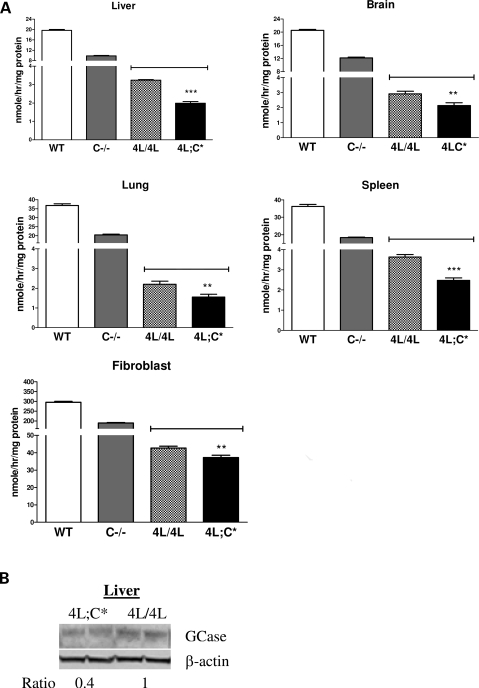
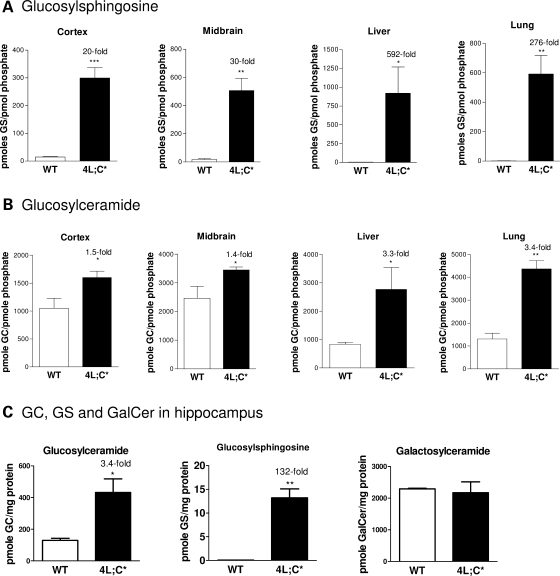
Gaucher disease is caused by defective acid beta-glucosidase (GCase) function. Saposin C is a lysosomal protein needed for optimal GCase activity. To test the in vivo effects of saposin C on GCase, saposin C deficient mice (C-/-) were backcrossed to point mutated GCase (V394L/V394L) mice. The resultant mice (4L;C*) began to exhibit CNS abnormalities approximately 30 days: first as hindlimb paresis, then progressive tremor and ataxia. Death occurred approximately 48 days due to neurological deficits. Axonal degeneration was evident in brain stem, spinal cord and white matter of cerebellum accompanied by increasing infiltration of the brain stem, cortex and thalamus by CD68 positive microglial cells and activation of astrocytes. Electron microscopy showed inclusion bodies in neuronal processes and degenerating cells. Accumulation of p62 and Lamp2 were prominent in the brain suggesting the impairment of autophagosome/lysosome function. This phenotype was different from either V394L/V394L or C-/- alone. Relative to V394L/V394L mice, 4L;C* mice had diminished GCase protein and activity. Marked increases (20- to 30-fold) of glucosylsphingosine (GS) and moderate elevation (1.5- to 3-fold) of glucosylceramide (GC) were in 4L;C* brains. Visceral tissues had increases of GS and GC, but no storage cells were found. Neuronal cells in thick hippocampal slices from 4L;C* mice had significantly attenuated long-term potentiation, presumably resulting from substrate accumulation. The 4L;C* mouse mimics the CNS phenotype and biochemistry of some type 3 (neuronopathic) variants of Gaucher disease and is a unique model suitable for testing pharmacological chaperone and substrate reduction therapies, and investigating the mechanisms of neuronopathic Gaucher disease.
Figures
Similar articles
-
Substrate compositional variation with tissue/region and Gba1 mutations in mouse models--implications for Gaucher disease.PLoS One. 2013;8(3):e57560. doi: 10.1371/journal.pone.0057560. Epub 2013 Mar 8. PLoS One. 2013. PMID: 23520473 Free PMC article.
-
Combination of acid β-glucosidase mutation and Saposin C deficiency in mice reveals Gba1 mutation dependent and tissue-specific disease phenotype.Sci Rep. 2019 Apr 3;9(1):5571. doi: 10.1038/s41598-019-41914-7. Sci Rep. 2019. PMID: 30944381 Free PMC article.
-
Gaucher disease mouse models: point mutations at the acid beta-glucosidase locus combined with low-level prosaposin expression lead to disease variants.J Lipid Res. 2005 Oct;46(10):2102-13. doi: 10.1194/jlr.M500202-JLR200. Epub 2005 Aug 1. J Lipid Res. 2005. PMID: 16061944
-
The role of saposin C in Gaucher disease.Mol Genet Metab. 2012 Jul;106(3):257-63. doi: 10.1016/j.ymgme.2012.04.024. Epub 2012 May 5. Mol Genet Metab. 2012. PMID: 22652185 Free PMC article. Review.
-
Molecular regulations and therapeutic targets of Gaucher disease.Cytokine Growth Factor Rev. 2018 Jun;41:65-74. doi: 10.1016/j.cytogfr.2018.04.003. Epub 2018 Apr 11. Cytokine Growth Factor Rev. 2018. PMID: 29699937 Free PMC article. Review.
Cited by
-
Targeting the Complement-Sphingolipid System in COVID-19 and Gaucher Diseases: Evidence for a New Treatment Strategy.Int J Mol Sci. 2022 Nov 18;23(22):14340. doi: 10.3390/ijms232214340. Int J Mol Sci. 2022. PMID: 36430817 Free PMC article. Review.
-
Developmental manganese neurotoxicity in rats: Cognitive deficits in allocentric and egocentric learning and memory.Neurotoxicol Teratol. 2017 Jan-Feb;59:16-26. doi: 10.1016/j.ntt.2016.10.005. Epub 2016 Oct 15. Neurotoxicol Teratol. 2017. PMID: 27756629 Free PMC article.
-
Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders.Int J Mol Sci. 2016 Jul 4;17(7):1065. doi: 10.3390/ijms17071065. Int J Mol Sci. 2016. PMID: 27384562 Free PMC article. Review.
-
A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis.Int J Mol Sci. 2021 May 28;22(11):5793. doi: 10.3390/ijms22115793. Int J Mol Sci. 2021. PMID: 34071409 Free PMC article. Review.
-
Progression of Behavioral and CNS Deficits in a Viable Murine Model of Chronic Neuronopathic Gaucher Disease.PLoS One. 2016 Sep 6;11(9):e0162367. doi: 10.1371/journal.pone.0162367. eCollection 2016. PLoS One. 2016. PMID: 27598339 Free PMC article.
References
-
- Beutler E., Grabowski G.A. In: The Metabolic and Molecular Basis of Inherited Disease. Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. Vol. III. New York: McGraw-Hill; 2001. pp. 3635–3668.
-
- Grabowski G.A., Kolodny E.H., Weinreb N.J., Rosenbloom B.E., Prakash-Cheng A., Kaplan P., Charrow J., Pastores G.M., Mistry P.K. In: The Metabolic and Molecular Bases of Inherited Diseases. Scriver C.R., Sly W.S., Beaudet A., Valle D., Childs B., editors. New York: McGraw-Hill; 2006.
-
- Sidransky E. Gaucher disease: complexity in a ‘simple’ disorder. Mol. Genet. Metab. 2004;83:6–15. - PubMed
-
- Orvisky E., Park J.K., Parker A., Walker J.M., Martin B.M., Stubblefield B.K., Uyama E., Tayebi N., Sidransky E. The identification of eight novel glucocerebrosidase (GBA) mutations in patients with Gaucher disease. Hum. Mutat. 2002;19:458–459. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
