

Viral induction of the zinc finger antiviral protein is IRF3-dependent but NF-kappaB-independent
- PMID: 20048147
- PMCID: PMC2825402
- DOI: 10.1074/jbc.M109.054486
Viral induction of the zinc finger antiviral protein is IRF3-dependent but NF-kappaB-independent
Abstract
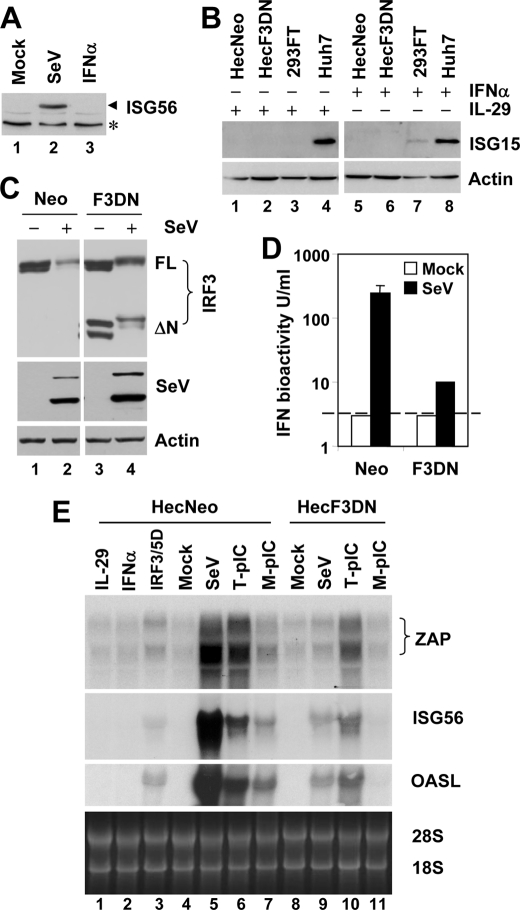
The zinc finger antiviral protein (ZAP) is an interferon-stimulated gene that restricts the replication of retroviruses, alphaviruses, and filoviruses. Relatively little is known, however, regarding the detailed mechanism of ZAP induction during viral infections. We show that, although being inducible by either interferon or virus, expression of ZAP is more efficiently activated by virus than are several other classical interferon-stimulated genes and that viral induction of ZAP occurs under the direct control of interferon regulatory factor 3 (IRF3) independent of interferon paracrine/autocrine signaling. ZAP was up-regulated in cells unresponsive to type I and III interferons upon engagement of TLR3, retinoic inducible gene I/melanoma differentiation-associated gene 5 pathways, or ectopic expression of a constitutively active IRF3 mutant. Conversely, induction of ZAP by virus or dsRNA was severely impaired in cells expressing a dominant-negative mutant IRF3 and completely abrogated in cells lacking IRF3. In contrast to IRF3, ZAP induction was independent of NF-kappaB activity. Mutational analysis of the human ZAP promoter revealed that multiple interferon-stimulated response elements far distal to the transcription start site serve redundantly to control IRF3-dependent induction of ZAP transcription. Chromatin immunoprecipitation assays demonstrated that IRF3 selectively binds the distal interferon-stimulated response elements in human ZAP promoter following viral infection. Collectively, these data suggest that ZAP is a direct target gene of IRF3 action in cellular antiviral responses.
Figures







Similar articles
-
Direct, interferon-independent activation of the CXCL10 promoter by NF-κB and interferon regulatory factor 3 during hepatitis C virus infection.J Virol. 2014 Feb;88(3):1582-90. doi: 10.1128/JVI.02007-13. Epub 2013 Nov 20. J Virol. 2014. PMID: 24257594 Free PMC article.
-
Differential Delivery of Genomic Double-Stranded RNA Causes Reovirus Strain-Specific Differences in Interferon Regulatory Factor 3 Activation.J Virol. 2018 Apr 13;92(9):e01947-17. doi: 10.1128/JVI.01947-17. Print 2018 May 1. J Virol. 2018. PMID: 29437975 Free PMC article.
-
Human Cytomegalovirus DNA Polymerase Subunit UL44 Antagonizes Antiviral Immune Responses by Suppressing IRF3- and NF-κB-Mediated Transcription.J Virol. 2019 May 15;93(11):e00181-19. doi: 10.1128/JVI.00181-19. Print 2019 Jun 1. J Virol. 2019. PMID: 30867312 Free PMC article.
-
VISA--a pass to innate immunity.Int J Biochem Cell Biol. 2007;39(2):287-91. doi: 10.1016/j.biocel.2006.08.016. Epub 2006 Sep 14. Int J Biochem Cell Biol. 2007. PMID: 17029998 Review.
-
Regulation of the MIR155 host gene in physiological and pathological processes.Gene. 2013 Dec 10;532(1):1-12. doi: 10.1016/j.gene.2012.12.009. Epub 2012 Dec 14. Gene. 2013. PMID: 23246696 Review.
Cited by
-
To translate, or not to translate: viral and host mRNA regulation by interferon-stimulated genes.Trends Cell Biol. 2015 Jun;25(6):320-9. doi: 10.1016/j.tcb.2015.02.001. Epub 2015 Mar 3. Trends Cell Biol. 2015. PMID: 25748385 Free PMC article. Review.
-
ZAP isoforms regulate unfolded protein response and epithelial- mesenchymal transition.Proc Natl Acad Sci U S A. 2022 Aug 2;119(31):e2121453119. doi: 10.1073/pnas.2121453119. Epub 2022 Jul 26. Proc Natl Acad Sci U S A. 2022. PMID: 35881805 Free PMC article.
-
Cell-to-Cell Variation in Defective Virus Expression and Effects on Host Responses during Influenza Virus Infection.mBio. 2020 Jan 14;11(1):e02880-19. doi: 10.1128/mBio.02880-19. mBio. 2020. PMID: 31937643 Free PMC article.
-
TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection.J Virol. 2011 Apr;85(8):3733-45. doi: 10.1128/JVI.02546-10. Epub 2011 Feb 2. J Virol. 2011. PMID: 21289118 Free PMC article.
-
Herpes simplex virus 1 UL41 protein abrogates the antiviral activity of hZAP by degrading its mRNA.Virol J. 2015 Dec 1;12:203. doi: 10.1186/s12985-015-0433-y. Virol J. 2015. PMID: 26625984 Free PMC article.
References
-
- Sen G. C. (2001) Annu. Rev. Microbiol. 55, 255–281 - PubMed
-
- Müller U., Steinhoff U., Reis L. F., Hemmi S., Pavlovic J., Zinkernagel R. M., Aguet M. (1994) Science 264, 1918–1921 - PubMed
-
- Hiscott J. (2007) J. Biol. Chem. 282, 15325–15329 - PubMed
-
- Osterlund P. I., Pietilä T. E., Veckman V., Kotenko S. V., Julkunen I. (2007) J. Immunol. 179, 3434–3442 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA140346/CA/NCI NIH HHS/United States
- U19-AI066316/AI/NIAID NIH HHS/United States
- R21-AI081058/AI/NIAID NIH HHS/United States
- R01 AI069285/AI/NIAID NIH HHS/United States
- U19 AI040035/AI/NIAID NIH HHS/United States
- R01 CA133322/CA/NCI NIH HHS/United States
- R01-AI069285/AI/NIAID NIH HHS/United States
- R21-DA018054/DA/NIDA NIH HHS/United States
- U19-AI40035/AI/NIAID NIH HHS/United States
- R21 AI081058/AI/NIAID NIH HHS/United States
- R21 DA018054/DA/NIDA NIH HHS/United States
- U19 AI066316/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
