

Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID
- PMID: 20048284
- PMCID: PMC2812546
- DOI: 10.1084/jem.20092238
Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID
Abstract
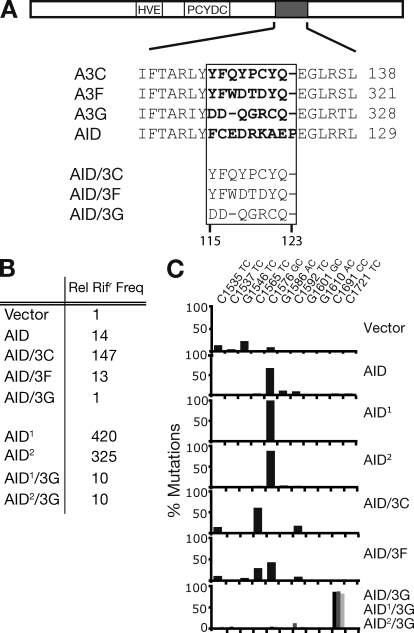
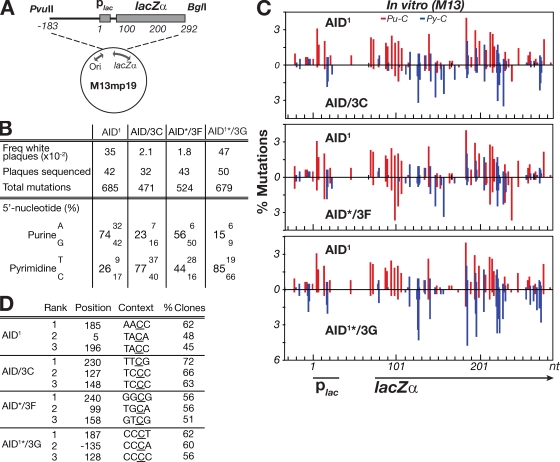
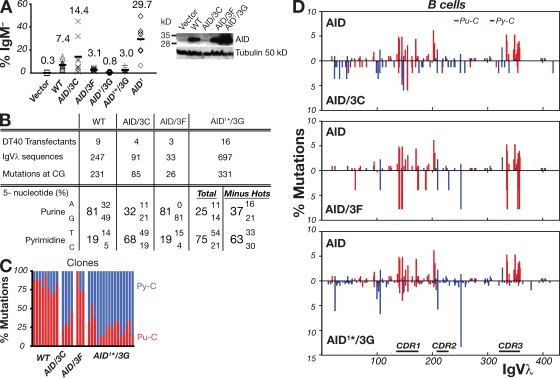
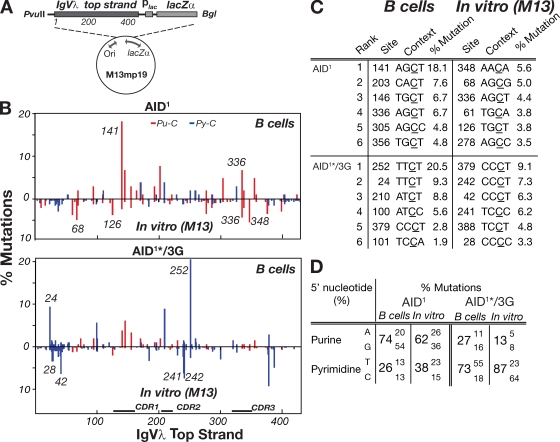
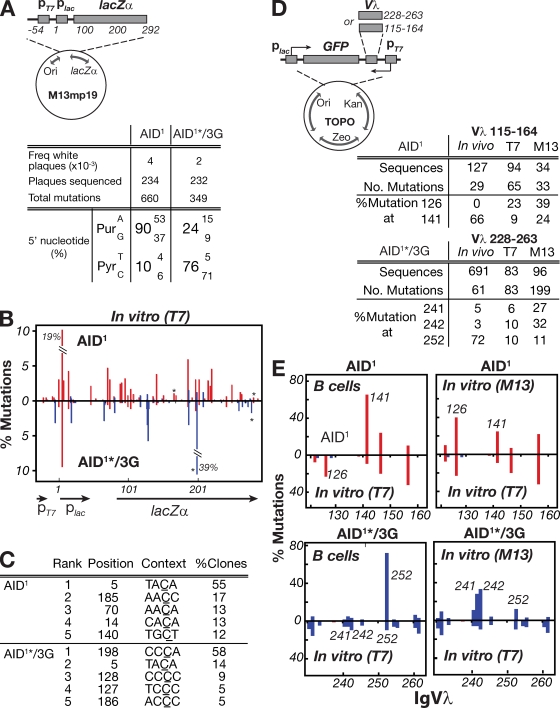
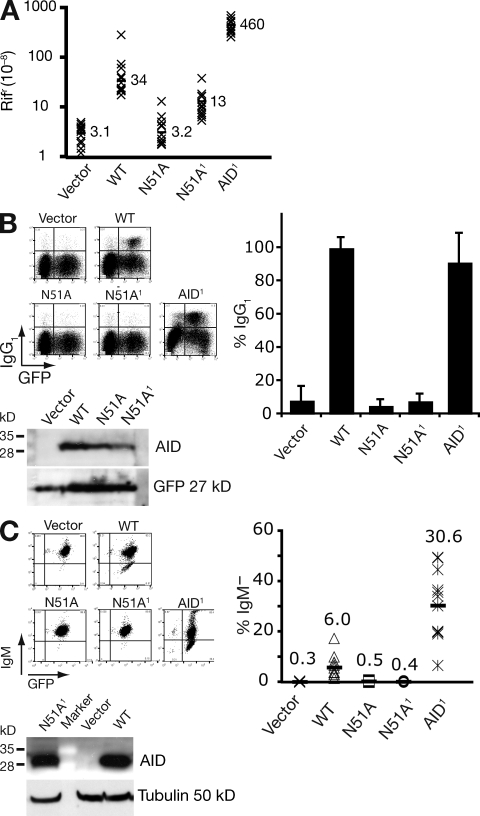
High-affinity antibodies are generated by somatic hypermutation with nucleotide substitutions introduced into the IgV in a semirandom fashion, but with intrinsic mutational hotspots strategically located to optimize antibody affinity maturation. The process is dependent on activation-induced deaminase (AID), an enzyme that can deaminate deoxycytidine in DNA in vitro, where its activity is sensitive to the identity of the 5'-flanking nucleotide. As a critical test of whether such DNA deamination activity underpins antibody diversification and to gain insight into the extent to which the antibody mutation spectrum is dependent on the intrinsic substrate specificity of AID, we investigated whether it is possible to change the IgV mutation spectrum by altering AID's active site such that it prefers a pyrimidine (rather than a purine) flanking the targeted deoxycytidine. Consistent with the DNA deamination mechanism, B cells expressing the modified AID proteins yield altered IgV mutation spectra (exhibiting a purine-->pyrimidine shift in flanking nucleotide preference) and altered hotspots. However, AID-catalyzed deamination of IgV targets in vitro does not yield the same degree of hotspot dominance to that observed in vivo, indicating the importance of features beyond AID's active site and DNA local sequence environment in determining in vivo hotspot dominance.
Figures
References
-
- Alt F.W., Honjo T. 2007. Advances in Immunology Vol. 94: AID for Immunoglobulin Diversity; Elsevier, Amsterdam
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
