

Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters
- PMID: 20048381
- PMCID: PMC3035512
- DOI: 10.1194/jlr.M003566
Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters
Abstract
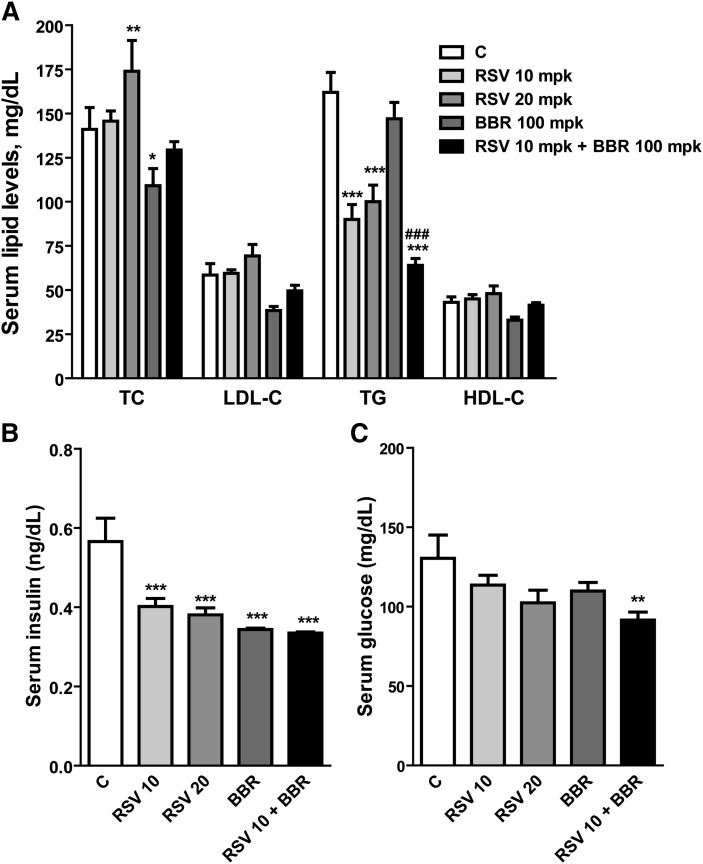
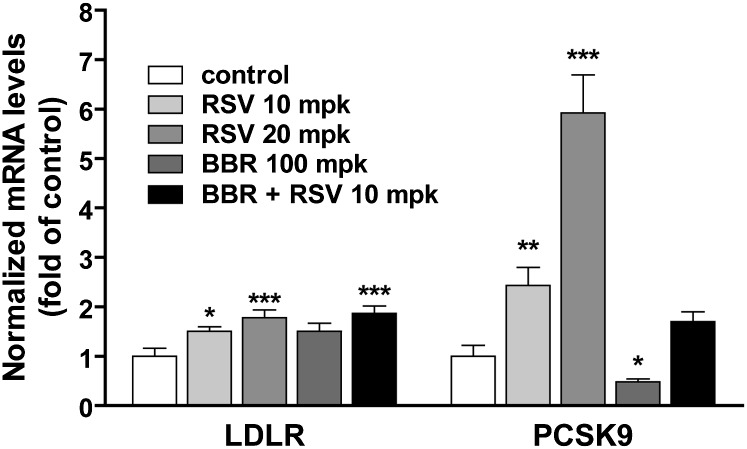
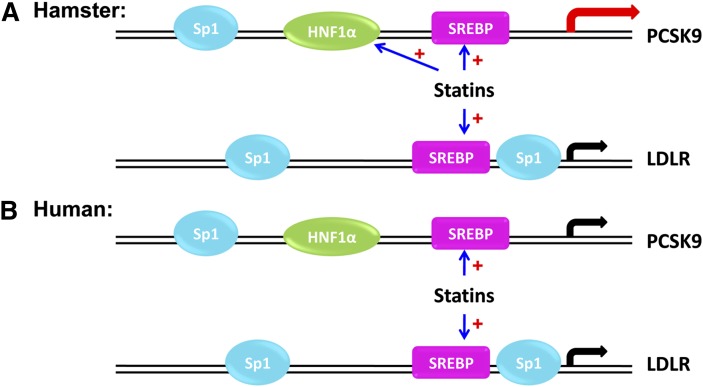
We investigated the role of proprotein convertase subtilisin/kexin type 9 (PCSK9) in the resistance of dyslipidemic hamsters to statin-induced LDL-cholesterol (LDL-C) reduction and the molecular mechanism by which statins modulated PCSK9 gene expression in vivo. We utilized the fructose diet-induced dyslipidemic hamsters as an in vivo model and rosuvastatin to examine its effects on liver PCSK9 and LDL receptor (LDLR) expression and serum lipid levels. We showed that rosuvastatin induced PCSK9 mRNA to a greater extent than LDLR mRNA in the hamster liver. The net result was that hepatic LDLR protein level was reduced. This correlated closely with an increase in serum LDL-C with statin treatment. More importantly, we demonstrated that in addition to an increase in sterol response element binding protein 2 (SREBP2) expression, rosuvastatin treatment increased the liver expression of hepatocyte nuclear factor 1 alpha (HNF1alpha), the newly identified key transactivator for PCSK9 gene expression. Our study suggests that the inducing effect of rosuvastatin on HNF1alpha is likely a underlying mechanism accounting for the higher induction of PCSK9 than LDLR because of the utilization of two transactivators (HNF1alpha and SREBP2) in PCSK9 transcription versus one (SREBP2) in LDLR transcription. Thus, the net balance is in favor of PCSK9-induced degradation of LDLR in the hamster liver, abrogating the effect of rosuvastatin on LDL-C lowering.
Figures




References
-
- Walsh K. M., Albassam M. A., Clarke D. E. 1996. Subchronic toxicity of atorvastatin, a hydroxymethylglutaryl-Coenzyme A reductase inhibitor, in beagle dogs. Toxicol. Pathol. 24: 468–476. - PubMed
-
- Madsen C. S., Janovitz E., Zhang R., Nguyen-Tran V., Ryan C. S., Yin X., Monshizadegan H., Chang M., D'Arienzo C., Scheer S., et al. 2008. The guinea pig as a preclinical model for demonstrating the efficacy and safety of statins. J. Pharmacol. Exp. Ther. 324: 576–586. - PubMed
-
- Ito B. R., Zhang B-H., Cable E. E., Song X., Fujitaki J. M., MacKenna D. A., Wilker C. E., Chi B., Poelje P. D., Linemeter D. L., et al. 2009. Thyroid hormone b receptor activation has additive cholesterol lowering activity in combination with atorvastatin in rabbits, dogs, and monkeys. B J Pharmacol. 156: 454–465. - PMC - PubMed
-
- Krause B. R., Newton R. S. 1995. Lipid-lowering activity of atorvastatin and lovastatin in rodent species: triglyceride-lowering in rats correlates with efficacy in LDL animal models. Atherosclerosis. 117: 237–244. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
