

A reexamination of the PPAR-alpha activation mode of action as a basis for assessing human cancer risks of environmental contaminants
- PMID: 20049115
- PMCID: PMC2801168
- DOI: 10.1289/ehp.0900758
A reexamination of the PPAR-alpha activation mode of action as a basis for assessing human cancer risks of environmental contaminants
Abstract
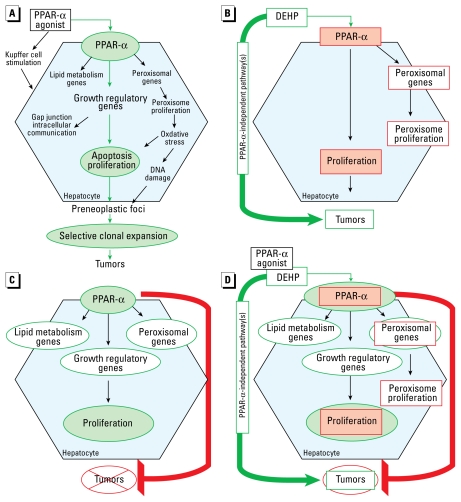
Background: Diverse environmental contaminants, including the plasticizer di(2-ethylhexyl)phthalate (DEHP), are hepatocarcinogenic peroxisome proliferators in rodents. Peroxisome proliferator-activated receptor-alpha (PPAR-alpha) activation and its sequelae have been proposed to constitute a mode of action (MOA) for hepatocarcinogenesis by such agents as a sole causative factor. Further, based on a hypothesized lower sensitivity of humans to this MOA, prior reviews have concluded that rodent hepatocarcinogenesis by PPAR-alpha agonists is irrelevant to human carcinogenic risk.
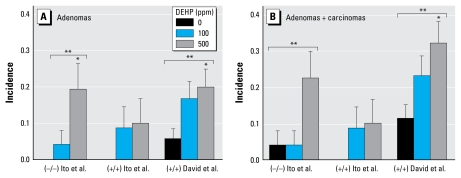
Data synthesis: Herein, we review recent studies that experimentally challenge the PPAR-alpha activation MOA hypothesis, providing evidence that DEHP is hepatocarcinogenic in PPAR-alpha-null mice and that the MOA but not hepatocarcinogenesis is evoked by PPAR-alpha activation in a transgenic mouse model. We further examine whether relative potency for PPAR-alpha activation or other steps in the MOA correlates with tumorigenic potency. In addition, for most PPAR-alpha agonists of environmental concern, available data are insufficient to characterize relative human sensitivity to this rodent MOA or to induction of hepatocarcinogenesis.
Conclusions: Our review and analyses raise questions about the hypothesized PPAR-alpha activation MOA as a sole explanation for rodent hepatocarcinogenesis by PPAR-alpha agonists and therefore its utility as a primary basis for assessing human carcinogenic risk from the diverse compounds that activate PPAR-alpha. These findings have broad implications for how MOA hypotheses are developed, tested, and applied in human health risk assessment. We discuss alternatives to the current approaches to these key aspects of mechanistic data evaluation.
Keywords: carcinogenesis; mode of action; peroxisome proliferators; risk assessment.
Figures
Similar articles
-
Mode of action framework analysis for receptor-mediated toxicity: The peroxisome proliferator-activated receptor alpha (PPARα) as a case study.Crit Rev Toxicol. 2014 Jan;44(1):1-49. doi: 10.3109/10408444.2013.835784. Epub 2013 Nov 4. Crit Rev Toxicol. 2014. PMID: 24180432 Review.
-
Rodent carcinogenicity of peroxisome proliferators and issues on human relevance.J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2004 May;22(1):37-55. doi: 10.1081/GNC-120038005. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2004. PMID: 15845221 Review.
-
Is peroxisome proliferation an obligatory precursor step in the carcinogenicity of di(2-ethylhexyl)phthalate (DEHP)?Environ Health Perspect. 2001 May;109(5):437-42. doi: 10.1289/ehp.01109437. Environ Health Perspect. 2001. PMID: 11401753 Free PMC article. Review.
-
A cancer risk assessment of di(2-ethylhexyl)phthalate: application of the new U.S. EPA Risk Assessment Guidelines.Regul Toxicol Pharmacol. 1999 Jun;29(3):327-57. doi: 10.1006/rtph.1999.1296. Regul Toxicol Pharmacol. 1999. PMID: 10388618 Review.
-
Characterization of peroxisome proliferator-activated receptor alpha--independent effects of PPARalpha activators in the rodent liver: di-(2-ethylhexyl) phthalate also activates the constitutive-activated receptor.Toxicol Sci. 2010 Jan;113(1):45-59. doi: 10.1093/toxsci/kfp251. Epub 2009 Oct 22. Toxicol Sci. 2010. PMID: 19850644 Free PMC article.
Cited by
-
The role of oxidative stress in carcinogenesis induced by metals and xenobiotics.Cancers (Basel). 2010 Apr 8;2(2):376-96. doi: 10.3390/cancers2020376. Cancers (Basel). 2010. PMID: 24281075 Free PMC article.
-
Occurrence of fibrates and their metabolites in source and drinking water in Shanghai and Zhejiang, China.Sci Rep. 2017 Apr 12;7:45931. doi: 10.1038/srep45931. Sci Rep. 2017. PMID: 28401920 Free PMC article.
-
Consensus on the key characteristics of endocrine-disrupting chemicals as a basis for hazard identification.Nat Rev Endocrinol. 2020 Jan;16(1):45-57. doi: 10.1038/s41574-019-0273-8. Epub 2019 Nov 12. Nat Rev Endocrinol. 2020. PMID: 31719706 Free PMC article. Review.
-
Plasticizers May Activate Human Hepatic Peroxisome Proliferator-Activated Receptor α Less Than That of a Mouse but May Activate Constitutive Androstane Receptor in Liver.PPAR Res. 2012;2012:201284. doi: 10.1155/2012/201284. Epub 2012 Jun 20. PPAR Res. 2012. PMID: 22792086 Free PMC article.
-
In vitro screening of environmental chemicals for targeted testing prioritization: the ToxCast project.Environ Health Perspect. 2010 Apr;118(4):485-92. doi: 10.1289/ehp.0901392. Environ Health Perspect. 2010. PMID: 20368123 Free PMC article.
References
-
- Ashby J, Brady A, Elcombe CR, Elliott BM, Ishmael J, Odum J, et al. Mechanistically-based human hazard assessment of peroxisome proliferator-induced hepatocarcinogenesis. Hum Exp Toxicol. 1994;13(suppl 2):S1–S117. - PubMed
-
- Barrass NC, Price RJ, Lake BG, Orton TC. Comparison of the acute and chronic mitogenic effects of the peroxisome proliferators methylclofenapate and clofibric acid in rat liver. Carcinogenesis. 1993;14(7):1451–1456. - PubMed
-
- Bezafibrate Infarction Prevention Study Group. Lipids and lipoproteins in symptomatic coronary heart disease. Distribution, intercorrelations, and significance for risk classification in 6,700 men and 1,500 women. Circulation. 1992;86(3):839–848. - PubMed
-
- Bezafibrate Infarction Prevention Study Group. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease: the Bezafibrate Infarction Prevention (BIP) study. Circulation. 2000;102(1):21–27. - PubMed
-
- Blumcke S, Schwartzkopff W, Lobeck H, Edmondson NA, Prentice DE, Blane GF. Influence of fenofibrate on cellular and subcellular liver structure in hyperlipidemic patients. Atherosclerosis. 1983;46(1):105–116. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
