

Characterization of proteins with wide-angle X-ray solution scattering (WAXS)
- PMID: 20049539
- PMCID: PMC3057577
- DOI: 10.1007/s10969-009-9075-x
Characterization of proteins with wide-angle X-ray solution scattering (WAXS)
Abstract
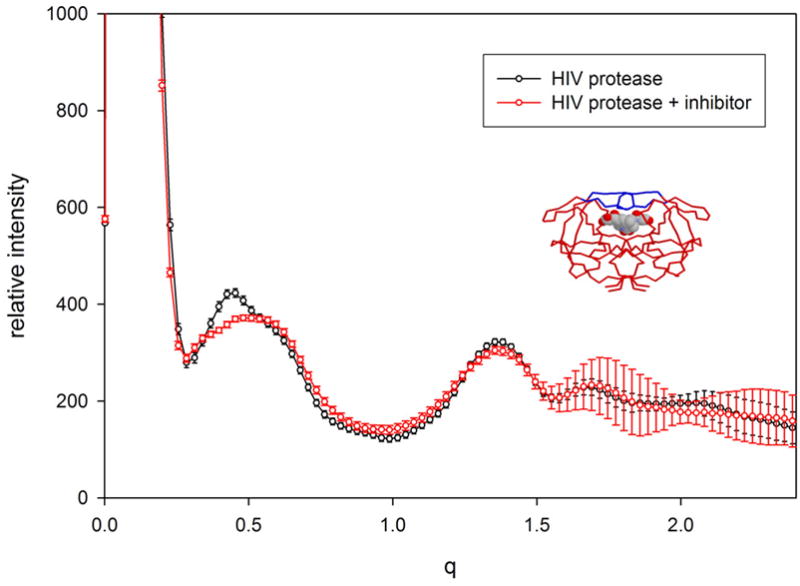
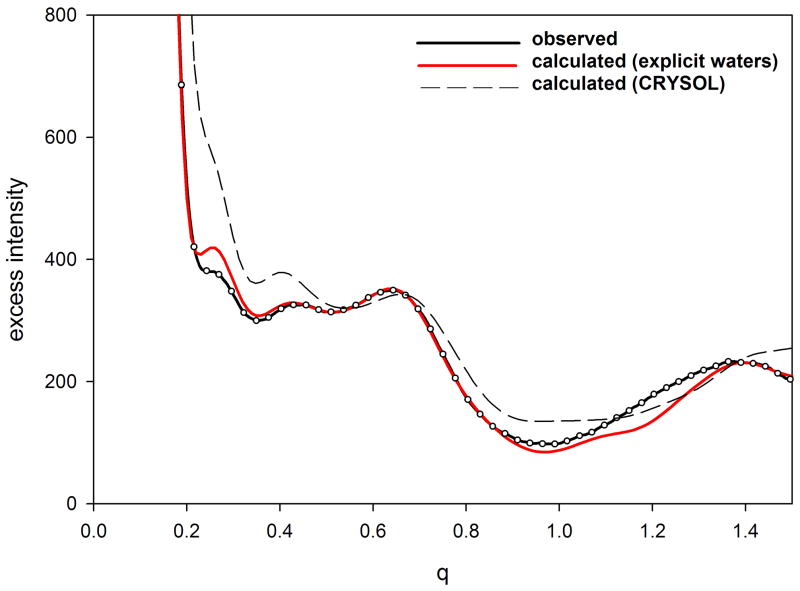
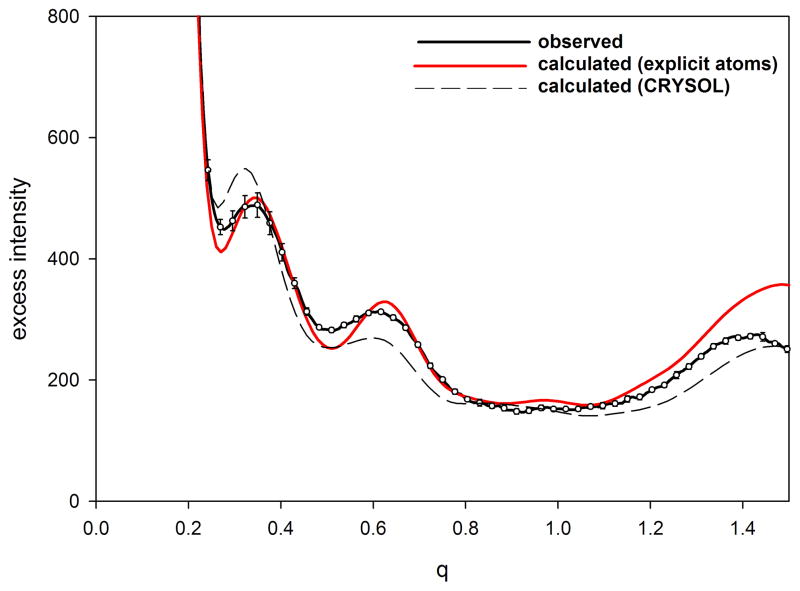
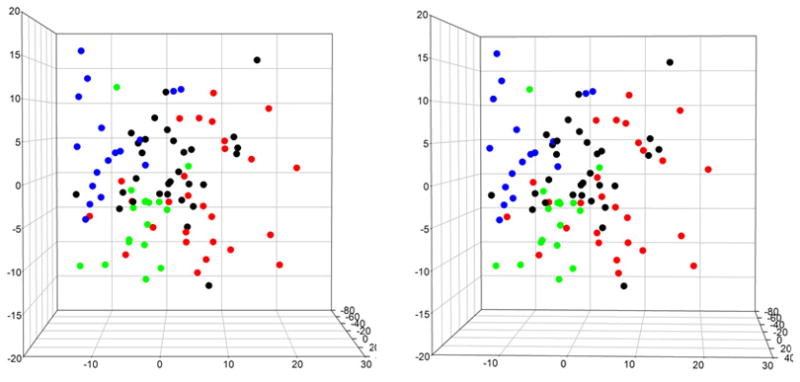
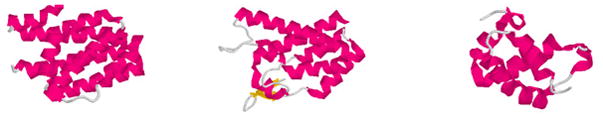
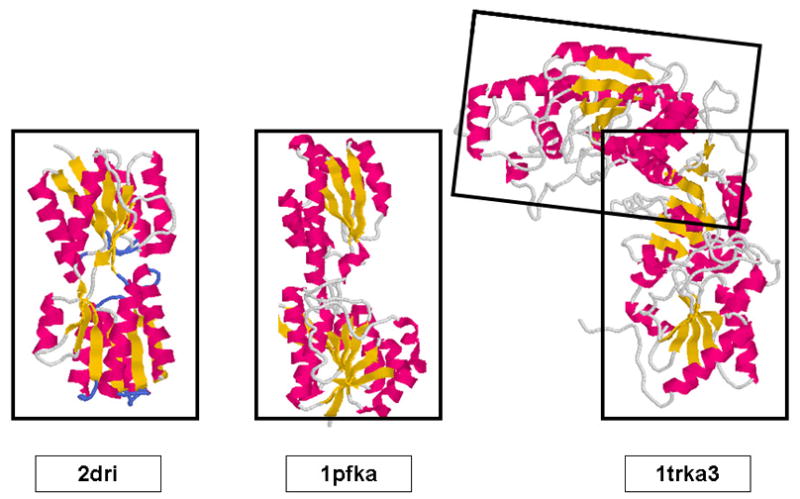
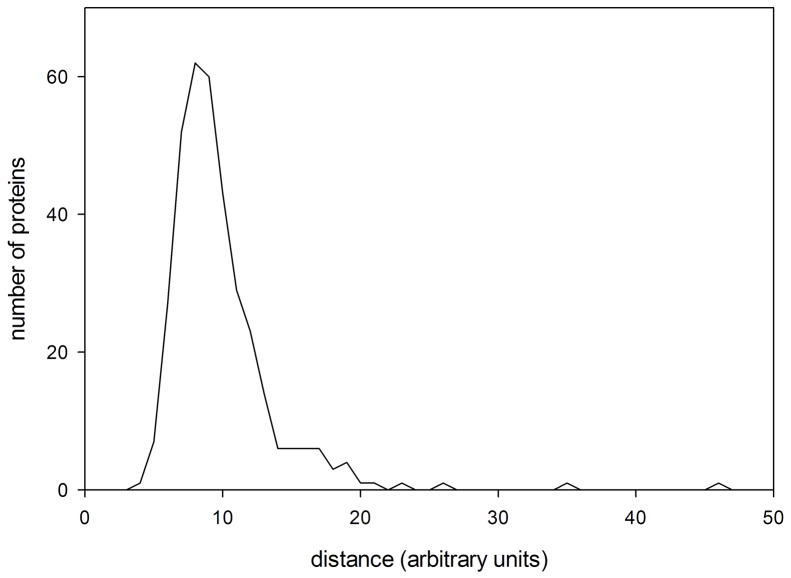
X-ray solution scattering in both the small-angle (SAXS) and wide-angle (WAXS) regimes is making an increasing impact on our understanding of biomolecular complexes. The accurate calculation of WAXS patterns from atomic coordinates has positioned the approach for rapid growth and integration with existing Structural Genomics efforts. WAXS data are sensitive to small structural changes in proteins; useful for calculation of the pair-distribution function at relatively high resolution; provides a means to characterize the breadth of the structural ensemble in solution; and can be used to identify proteins with similar folds. WAXS data are often used to test structural models, identify structural similarities and characterize structural changes. WAXS is highly complementary to crystallography and NMR. It holds great potential for the testing of structural models of proteins; identification of proteins that may exhibit novel folds; characterization of unfolded or natively disordered proteins; and detection of structural changes associated with protein function.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
