

The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease
- PMID: 20049702
- PMCID: PMC3378108
- DOI: 10.1002/emmm.200900002
The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease
Abstract
A novel class of antibiotic acyldepsipeptides (designated ADEPs) exerts its unique antibacterial activity by targeting the peptidase caseinolytic protease P (ClpP). ClpP forms proteolytic complexes with heat shock proteins (Hsp100) that select and process substrate proteins for ClpP-mediated degradation. Here, we analyse the molecular mechanism of ADEP action and demonstrate that ADEPs abrogate ClpP interaction with cooperating Hsp100 adenosine triphosphatases (ATPases). Consequently, ADEP treated bacteria are affected in ClpP-dependent general and regulatory proteolysis. At the same time, ADEPs also activate ClpP by converting it from a tightly regulated peptidase, which can only degrade short peptides, into a proteolytic machinery that recognizes and degrades unfolded polypeptides. In vivo nascent polypeptide chains represent the putative primary target of ADEP-activated ClpP, providing a rationale for the antibacterial activity of the ADEPs. Thus, ADEPs cause a complete functional reprogramming of the Clp-protease complex.
Figures
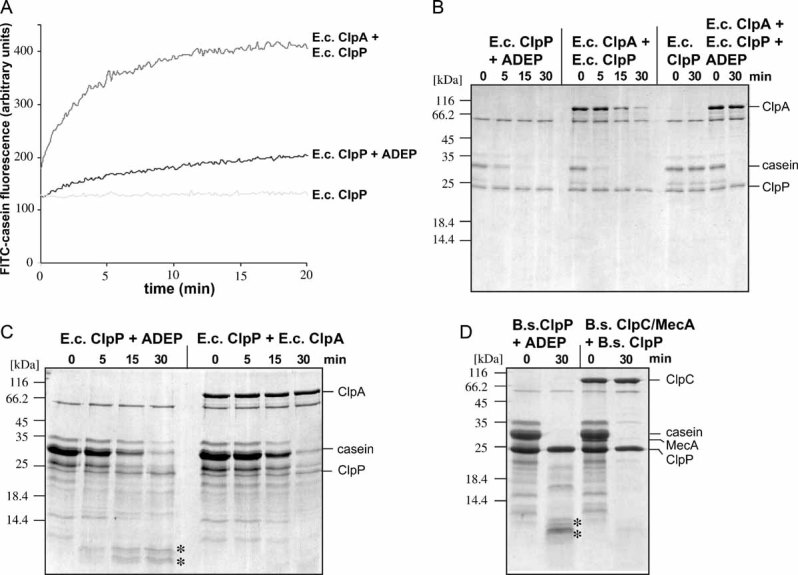
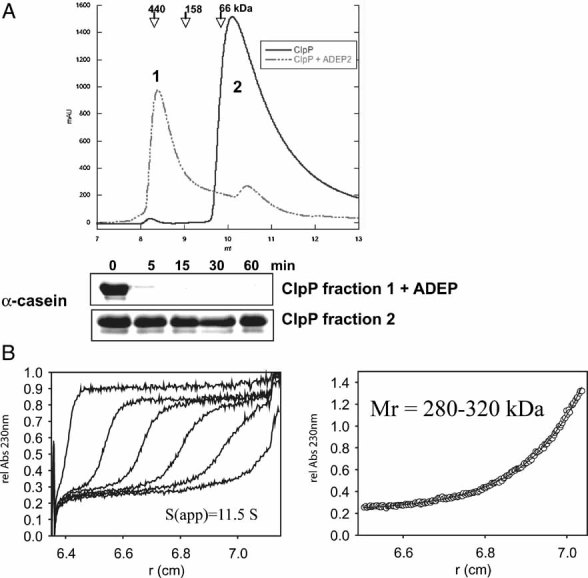
The oligomeric state of ClpP in the absence (solid line) and presence (dashed line) of ADEP (0.5 µg/ml) was monitored by size exclusion chromatography using a Superdex 75 column. Elution positions of protein standards are given. Samples of both eluted peak fractions (as indicated in the chromatogram: 1: ClpP oligomer and 2: ClpP monomer) were incubated with 1 µM casein and tested for proteolytic activity by SDS–PAGE analysis.
The association state of 10 µM ClpP in the presence of 12 µM ADEP was analysed by analytical ultracentrifugation. The left panel shows the sedimentation velocity at 40,000 rpm and 20 °C (scans taken after every 20 min intervals depicted). The calculated sedimentation velocity was s(app) = 11.5 S. The right panel displays the data of a sedimentation equilibrium experiment after 60 h at 6,000 rpm and 20 °C. In several independent experiments, the molecular mass of ClpP was determined over the range of 280–320 kDa, fitting well to the size of a double heptameric structure of ClpP. The offset of A230 nm = 0.23 in both sedimentation velocity and equilibrium experiments arises from non-bound ADEP, which has been used in a slight molar excess over ClpP.
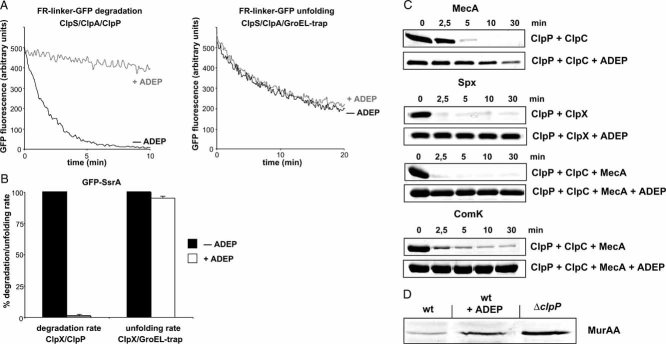
Degradation and unfolding of the N-end rule model substrate FR-linker-GFP (100 nM) was followed by monitoring the decrease of GFP fluorescence in the presence of the indicated components (+/− ADEP).
Degradation and unfolding of GFP-SsrA (100 nM) was followed by monitoring the decrease of GFP fluorescence in the presence of the indicated components (+/− ADEP). The rate of GFP-SsrA degradation and unfolding determined in the absence of ADEP was set as 100%.
In vitro degradation of MecA by ClpC/ClpP and ComK by ClpC/ClpP/MecA and Spx by either ClpC/ClpP/MecA or ClpX/ClpP in the absence and presence of ADEP. Substrate degradation was analysed by SDS–PAGE (all proteins were used at 1 µM).
Analysis of the in vivo stability of MurAA in B. subtilis cells (wild type (wt) and ΔclpP mutant). ADEP (final concentration: 1.6 µg/ml) was added upon entry of cells into stationary phase and cells were incubated for an additional period of 2 h prior to harvesting. MurAA levels were determined by immunoblot analysis using MurAA-specific antibodies.
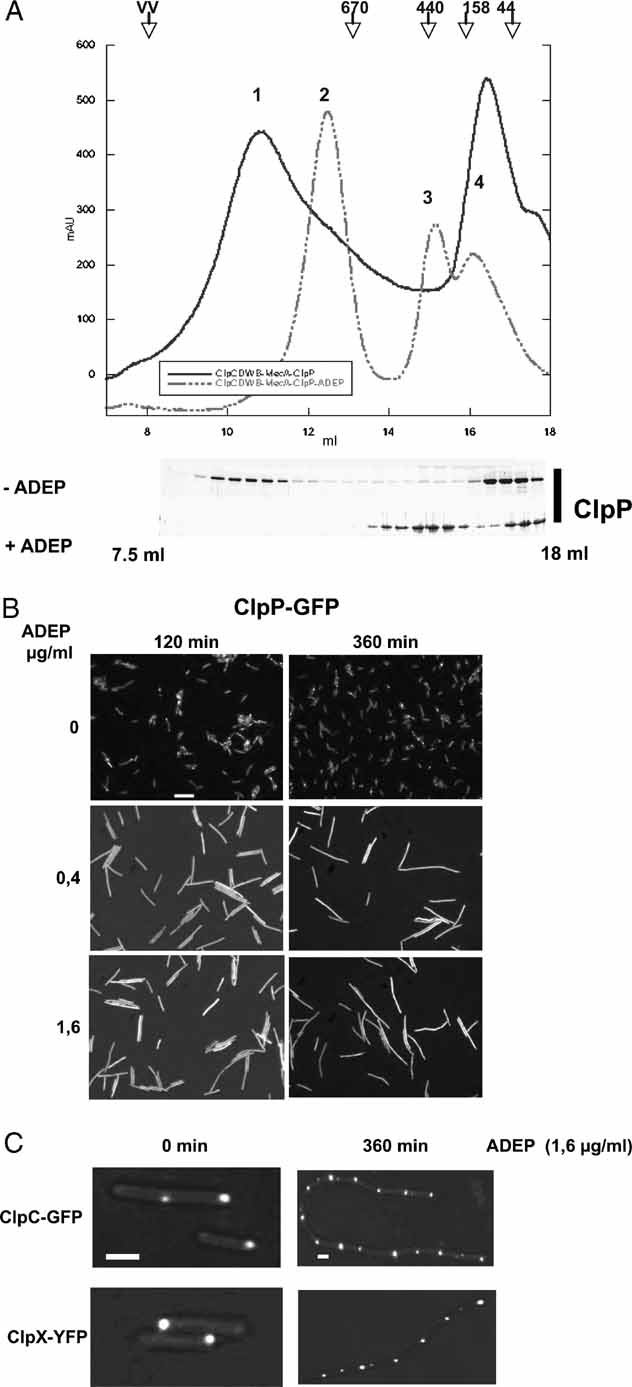
A complex of ClpC-DWB, MecA and ClpP was preformed in the presence of 5 mM ATP at 37 °C for 10 min. ADEP (0.5 µg/ml) was subsequently added and the sample was further incubated for an additional 10 min. As a control, a sample without the addition of ADEP was analysed in parallel. The association and dissociation of the respective complexes were analysed by size exclusion chromatography using a Superose 6 column. The chromatogram of ClpC-DWB, MecA and ClpP is shown by a solid line and the chromatogram of the ADEP including sample by a dashed line. The resulting peaks (as indicated in the chromatogram) could be assigned as follows: 1: ClpC-DWB6MecA6/(ClpP7)2/ClpC-DWB6MecA6; 2: ClpC-DWB6/MecA6; 3: (ClpP7)2-ADEP and 4: ClpC_DWB/MecA heterodimer. The elution profile of ClpP is shown for both samples as a Coomassie-stained SDS gel. Elution positions of protein standards are given.
ADEP causes a delocalization of ClpP in bacterial cells. Localization of ClpP-GFP in B. subtilis 168 in the absence (upper row) or presence of ADEP (0.4 and 1.6 µg/ml middle and bottom row, respectively) is shown. ADEP was added during the logarithmic growth phase (30 °C, OD600nm = 1) and ClpP-GFP localization was monitored 120 and 360 min after ADEP addition by fluorescence microscopy. A scale bar of 5 µm length is depicted.
ADEP does not affect ClpC and ClpX localization in bacterial cells. Localization of ClpC-GFP and ClpX-YFP in B. subtilis 168 in the absence or presence of ADEP. Cells were treated as described in (B) and localization of fluorescent fusion proteins was analysed prior to and 360 min after the addition of ADEP. Scale bars are depicted for the left (1 µm) and right columns (2 µm).
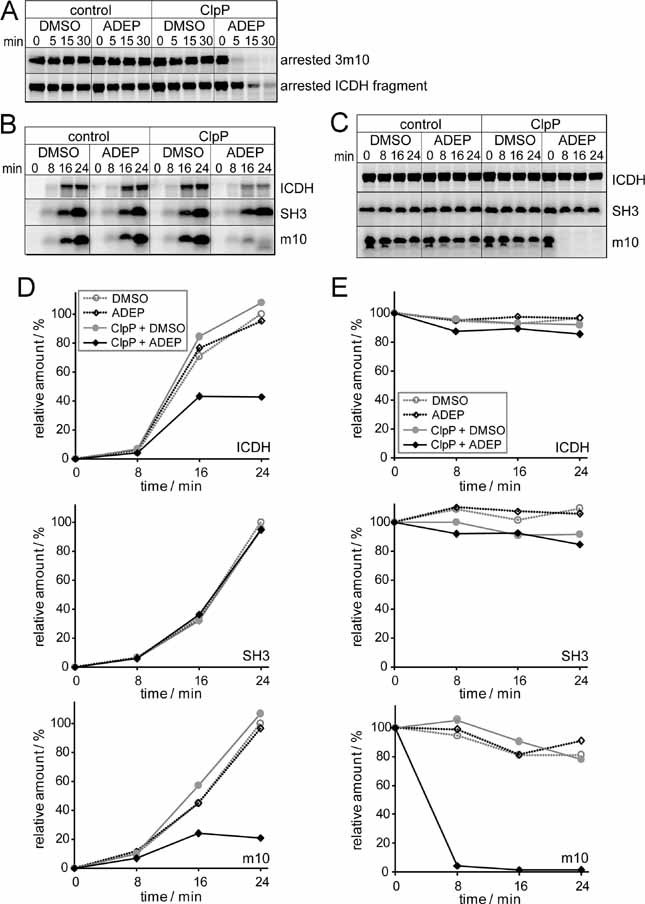
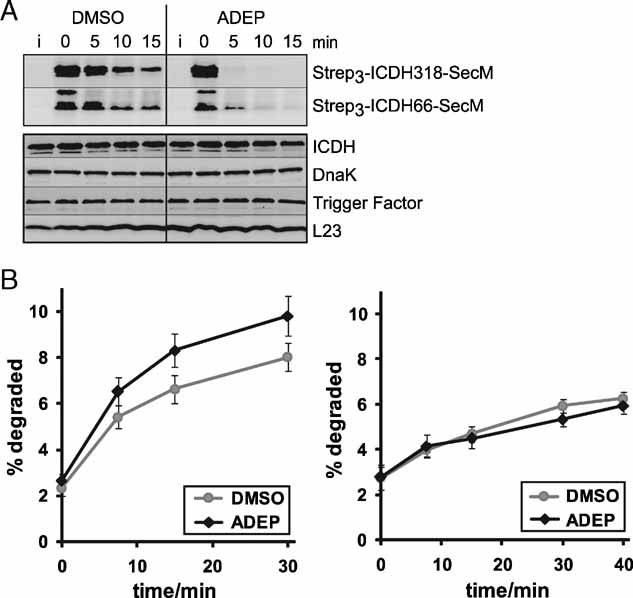
ADEP causes the degradation of ribosome-arrested nascent polypeptides in vivo. The degradation of two ribosome-arrested nascent chains derived from ICDH (Strep3-ICDH66-SecM, Strep3-ICDH318-SecM) was monitored in ER2566 ΔacrA::kan cells at 37 °C. Two hours after induction (i) of the model constructs, translation was stopped with chloramphenicol and ADEP or DMSO was added (0 min). At the indicated time points, cells were harvested and proteins separated by SDS–PAGE followed by Western blots against the N-terminal Strep-tag (upper panel) and against ICDH, DnaK, Trigger Factor and L23 as controls (lower panel). Only full-length products are depicted.
The degradation of newly synthesized proteins is increased in the presence of ADEP in vivo. Total protein degradation was determined in HN818 ΔacrA::kan cells in the exponential phase at 37 °C. ADEP1 and DMSO were either added before (left panel) or after (right panel) [35S] methionine pulse labelling. At the denoted time points, aliquots were TCA precipitated and the radioactivity in the TCA-soluble and insoluble fractions was determined by scintillation counting. The amount of degradation is given as percentage of the total cellular radioactivity. Mean values and standard errors of the mean of four to five independent experiments are shown.
References
-
- Blanco FJ, Angrand I, Serrano L. Exploring the conformational properties of the sequence space between two proteins with different folds: an experimental study. J Mol Biol. 1999;285:741–753. - PubMed
-
- Brötz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K, Bandow JE, Sahl HG, Labischinski H. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med. 2005;11:1082–1087. - PubMed
-
- Bukau B, Weissman J, Horwich AL. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
