

Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics
- PMID: 20049730
- PMCID: PMC3378140
- DOI: 10.1002/emmm.200900036
Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics
Abstract
Lysosomal storage diseases (LSDs) are a group of genetic disorders due to defects in any aspect of lysosomal biology. During the past two decades, different approaches have been introduced for the treatment of these conditions. Among them, enzyme replacement therapy (ERT) represented a major advance and is used successfully in the treatment of some of these disorders. However, ERT has limitations such as insufficient biodistribution of recombinant enzymes and high costs. An emerging strategy for the treatment of LSDs is pharmacological chaperone therapy (PCT), based on the use of chaperone molecules that assist the folding of mutated enzymes and improve their stability and lysosomal trafficking. After proof-of-concept studies, PCT is now being translated into clinical applications for Fabry, Gaucher and Pompe disease. This approach, however, can only be applied to patients carrying chaperone-responsive mutations. The recent demonstration of a synergistic effect of chaperones and ERT expands the applications of PCT and prompts a re-evaluation of their therapeutic use and potential. This review discusses the strengths and drawbacks of the potential therapies available for LSDs and proposes that future research should be directed towards the development of treatment protocols based on the combination of different therapies to improve the clinical outcome of LSD patients.
Figures
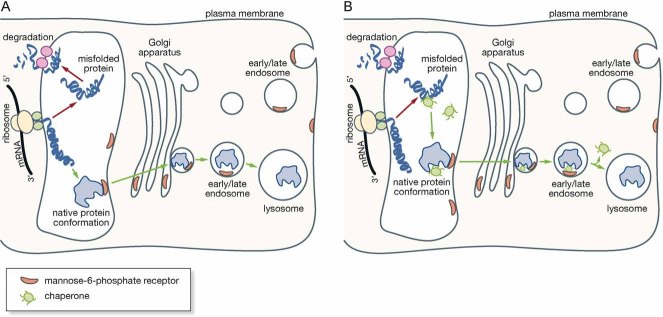
Folding of lysosomal enzymes is assisted by endogenous molecular chaperones. While wild type enzymes are properly folded by the chaperones and correctly transported to their destination (green arrows), mutated enzymes fail to fold efficiently into their native conformation, and are retro-translocated into the cytosol and degraded by the ERAD machinery (red arrows).
Pharmacological chaperones favour the folding of mutated enzymes that retain their catalytic activity, and prevent their recognition by the quality control system. The complex chaperone-mutated enzyme is delivered, at least partially, to the lysosomal compartment, where the chaperone is displaced from the active site of the enzyme by the excess natural substrate.
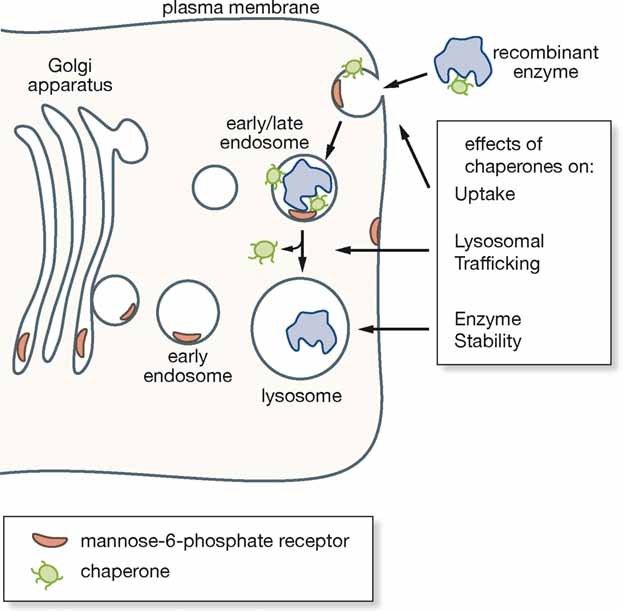
By improving the uptake of the recombinant enzymes by mutant cells,
Favouring their intracellular trafficking to lysosomes and
Increasing the enzyme's stability. Improved lysosomal trafficking is an important therapeutic goal, as these organelles are the site of substrate storage and because in the late endosomal/lysosomal compartment enzymes are processed into the mature and active isoforms. Increased stability may result in sustained corrective levels of intracellular enzymes.
Similar articles
-
Personalized Pharmacoperones for Lysosomal Storage Disorder: Approach for Next-Generation Treatment.Adv Protein Chem Struct Biol. 2016;102:225-65. doi: 10.1016/bs.apcsb.2015.10.001. Epub 2015 Nov 26. Adv Protein Chem Struct Biol. 2016. PMID: 26827607 Review.
-
Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders.Mol Ther. 2015 Jul;23(7):1138-1148. doi: 10.1038/mt.2015.62. Epub 2015 Apr 16. Mol Ther. 2015. PMID: 25881001 Free PMC article. Review.
-
Recent developments in therapeutic approaches for lysosomal storage diseases.Recent Pat CNS Drug Discov. 2011 Jan;6(1):1-19. doi: 10.2174/157488911794079127. Recent Pat CNS Drug Discov. 2011. PMID: 21073432 Review.
-
Therapeutic Role of Pharmacological Chaperones in Lysosomal Storage Disorders: A Review of the Evidence and Informed Approach to Reclassification.Biomolecules. 2023 Aug 7;13(8):1227. doi: 10.3390/biom13081227. Biomolecules. 2023. PMID: 37627292 Free PMC article. Review.
-
Treatment of lysosomal storage diseases: recent patents and future strategies.Recent Pat Endocr Metab Immune Drug Discov. 2014 Jan;8(1):9-25. doi: 10.2174/1872214808666140115111350. Recent Pat Endocr Metab Immune Drug Discov. 2014. PMID: 24433521 Review.
Cited by
-
Delivery and tracking of quantum dot peptide bioconjugates in an intact developing avian brain.ACS Chem Neurosci. 2015 Mar 18;6(3):494-504. doi: 10.1021/acschemneuro.5b00022. Epub 2015 Mar 5. ACS Chem Neurosci. 2015. PMID: 25688887 Free PMC article.
-
Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders.Assay Drug Dev Technol. 2011 Jun;9(3):213-35. doi: 10.1089/adt.2011.0370. Assay Drug Dev Technol. 2011. PMID: 21612550 Free PMC article. Review.
-
In Vitro Enzyme Measurement to Test Pharmacological Chaperone Responsiveness in Fabry and Pompe Disease.J Vis Exp. 2017 Dec 20;(130):56550. doi: 10.3791/56550. J Vis Exp. 2017. PMID: 29286471 Free PMC article.
-
Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant β-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease.Sci Rep. 2015 Jun 5;5:10903. doi: 10.1038/srep10903. Sci Rep. 2015. PMID: 26045184 Free PMC article.
-
β-Glucosidase 2 (GBA2) activity and imino sugar pharmacology.J Biol Chem. 2013 Sep 6;288(36):26052-26066. doi: 10.1074/jbc.M113.463562. Epub 2013 Jul 23. J Biol Chem. 2013. PMID: 23880767 Free PMC article.
References
-
- Andersson U, Smith D, Jeyakumar M, Butters TD, Borja MC, Dwek RA, Platt FM. Improved outcome of N-butyldeoxygalactonojirimycin-mediated substrate reduction therapy in a mouse model of Sandhoff disease. Neurobiol Dis. 2004;16:506–515. - PubMed
-
- Arakawa T, Ejima D, Kita Y, Tsumoto K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim Biophys Acta. 2006;1764:1677–1687. - PubMed
-
- Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793:684–696. Epub 2008 Dec 8. - PubMed
-
- Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, Mankin HJ, Murray GJ, Parker RI, Argoff CE, et al. Replacement therapy for inherited enzyme deficiency–macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991;324:1464–1470. - PubMed
-
- Benjamin ER, Flanagan JJ, Schilling A, Chang HH, Agarwal L, Katz E, Wu X, Pine C, Wustman B, Desnick RJ, et al. The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. J Inherit Metab Dis. 2009;32:424–440. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
