

Inactivation of the tumour suppressor, PTEN, in smooth muscle promotes a pro-inflammatory phenotype and enhances neointima formation
- PMID: 20051384
- PMCID: PMC2856191
- DOI: 10.1093/cvr/cvp425
Inactivation of the tumour suppressor, PTEN, in smooth muscle promotes a pro-inflammatory phenotype and enhances neointima formation
Abstract
Aims: Phosphatase and tensin homolog (PTEN) is implicated as a negative regulator of vascular smooth muscle cell (SMC) proliferation and injury-induced vascular remodelling. We tested if selective depletion of PTEN only in SMC is sufficient to promote SMC phenotypic modulation, cytokine production, and enhanced neointima formation.
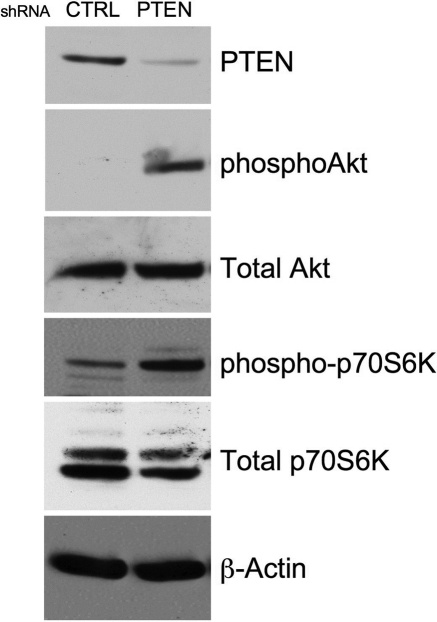
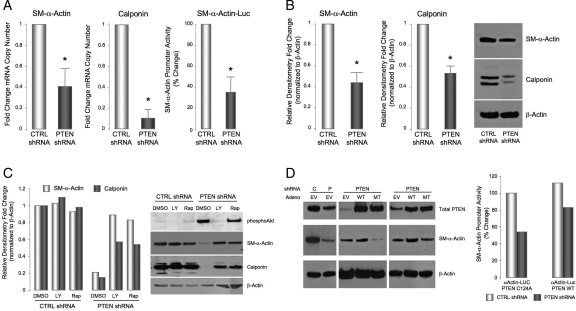
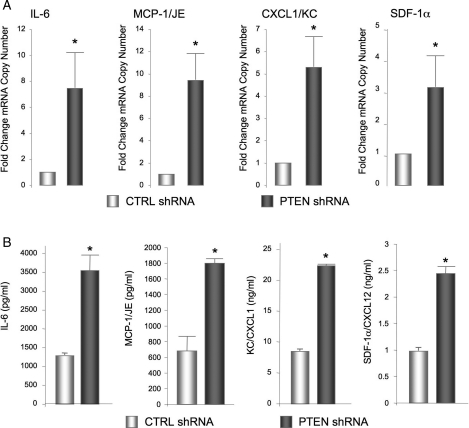
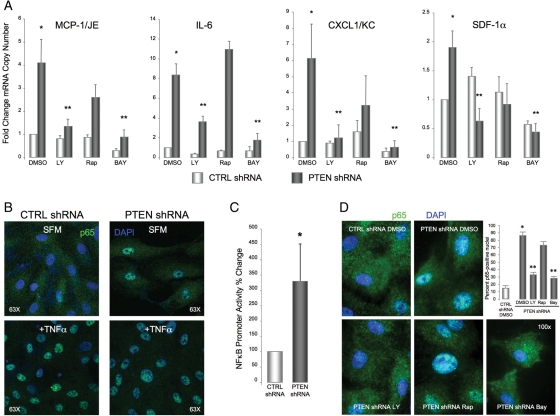
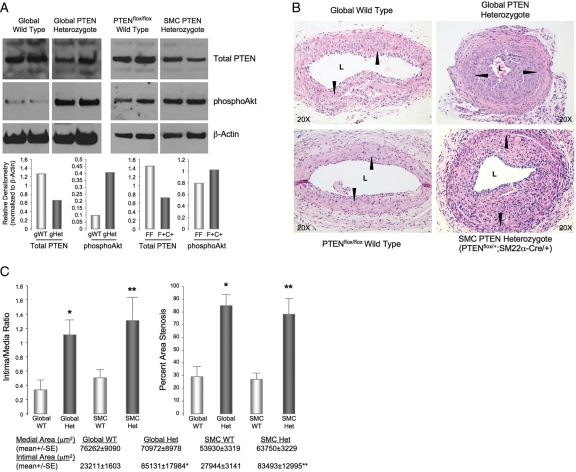
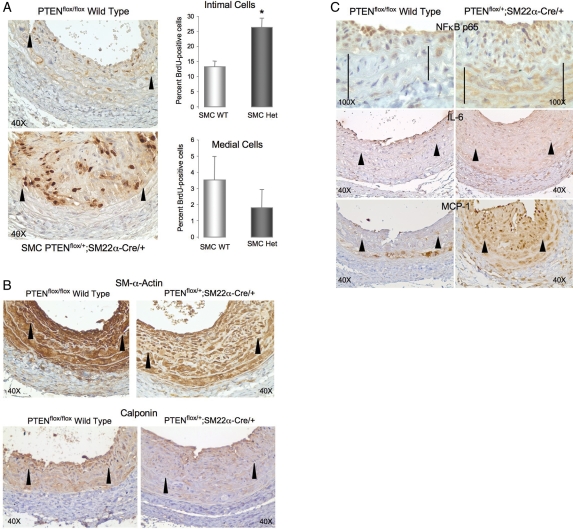
Methods and results: Smooth muscle marker expression and induction of pro-inflammatory cytokines were compared in cultured SMC expressing control or PTEN-specific shRNA. Compared with controls, PTEN-deficient SMC exhibited increased phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signalling and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kappaB) activity, reduced expression of SM markers (SM-alpha-actin and calponin), and increased production of stromal cell-derived factor-1alpha (SDF-1alpha), monocyte chemotactic protein-1 (MCP-1), interleukin-6 (IL-6), and chemokine (C-X-C motif) ligand 1 (KC/CXCL1) under basal conditions. PI3K/Akt or mTOR inhibition reversed repression of SM marker expression, whereas PI3K/Akt or NF-kappaB inhibition blocked cytokine induction mediated by PTEN depletion. Carotid ligation in mice with genetic reduction of PTEN specifically in SMC (SMC-specific PTEN heterozygotes) resulted in enhanced neointima formation, increased SMC hyperplasia, reduced SM-alpha-actin and calponin expression, and increased NF-kappaB and cytokine expression compared with wild-types. Lesion formation in SMC-specific heterozygotes was similar to lesion formation in global PTEN heterozygotes, indicating that inactivation of PTEN exclusively in SMC is sufficient to induce considerable increases in neointima formation.
Conclusion: PTEN activation specifically in SMC is a common upstream regulator of multiple downstream events involved in pathological vascular remodelling, including proliferation, de-differentiation, and production of multiple cytokines.
Figures
Similar articles
-
Targeted deletion of PTEN in smooth muscle cells results in vascular remodeling and recruitment of progenitor cells through induction of stromal cell-derived factor-1alpha.Circ Res. 2008 May 9;102(9):1036-45. doi: 10.1161/CIRCRESAHA.107.169896. Epub 2008 Mar 13. Circ Res. 2008. PMID: 18340011
-
The mechanical stress-activated serum-, glucocorticoid-regulated kinase 1 contributes to neointima formation in vein grafts.Circ Res. 2010 Nov 12;107(10):1265-74. doi: 10.1161/CIRCRESAHA.110.222588. Epub 2010 Sep 30. Circ Res. 2010. PMID: 20884880
-
CKII-SIRT1-SM22α loop evokes a self-limited inflammatory response in vascular smooth muscle cells.Cardiovasc Res. 2017 Aug 1;113(10):1198-1207. doi: 10.1093/cvr/cvx048. Cardiovasc Res. 2017. PMID: 28419207
-
Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis.J Vasc Res. 2010;47(2):168-80. doi: 10.1159/000250095. Epub 2009 Oct 22. J Vasc Res. 2010. PMID: 19851078 Free PMC article. Review.
-
Transcriptional regulation of vascular smooth muscle cell proliferation, differentiation and senescence: Novel targets for therapy.Vascul Pharmacol. 2022 Oct;146:107091. doi: 10.1016/j.vph.2022.107091. Epub 2022 Jul 25. Vascul Pharmacol. 2022. PMID: 35896140 Review.
Cited by
-
PTEN: an emerging target in rheumatoid arthritis?Cell Commun Signal. 2024 Apr 26;22(1):246. doi: 10.1186/s12964-024-01618-6. Cell Commun Signal. 2024. PMID: 38671436 Free PMC article. Review.
-
Potential Target miR-455 Delaying Arterial Stenosis Progression Through PTEN.Front Cardiovasc Med. 2021 Feb 23;8:611116. doi: 10.3389/fcvm.2021.611116. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 33708803 Free PMC article.
-
Activation of the retinoid X receptor modulates angiotensin II-induced smooth muscle gene expression and inflammation in vascular smooth muscle cells.Mol Pharmacol. 2014 Nov;86(5):570-9. doi: 10.1124/mol.114.092163. Epub 2014 Aug 28. Mol Pharmacol. 2014. PMID: 25169989 Free PMC article.
-
Small RNA sequencing reveals microRNAs that modulate angiotensin II effects in vascular smooth muscle cells.J Biol Chem. 2012 May 4;287(19):15672-83. doi: 10.1074/jbc.M111.322669. Epub 2012 Mar 19. J Biol Chem. 2012. PMID: 22431733 Free PMC article.
-
Minor introns are embedded molecular switches regulated by highly unstable U6atac snRNA.Elife. 2013 Jul 30;2:e00780. doi: 10.7554/eLife.00780. Elife. 2013. PMID: 23908766 Free PMC article.
References
-
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. - PubMed
-
- Glass CK, Witztum JL. Atherosclerosis, the road ahead. Cell. 2001;104:503–516. - PubMed
-
- Schober A. Chemokines in vascular dysfunction and remodeling. Arterioscler Thromb Vasc Biol. 2008;28:1950–1959. - PubMed
-
- Taubman MB, Rollins BJ, Poon M, Marmur J, Green RS, Berk BC, et al. JE mRNA accumulates rapidly in aortic injury and in platelet-derived growth factor-stimulated vascular smooth muscle cells. Circ Res. 1992;70:314–325. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
