

Multiscale simulations of protein landscapes: using coarse-grained models as reference potentials to full explicit models
- PMID: 20052756
- PMCID: PMC2822134
- DOI: 10.1002/prot.22640
Multiscale simulations of protein landscapes: using coarse-grained models as reference potentials to full explicit models
Abstract
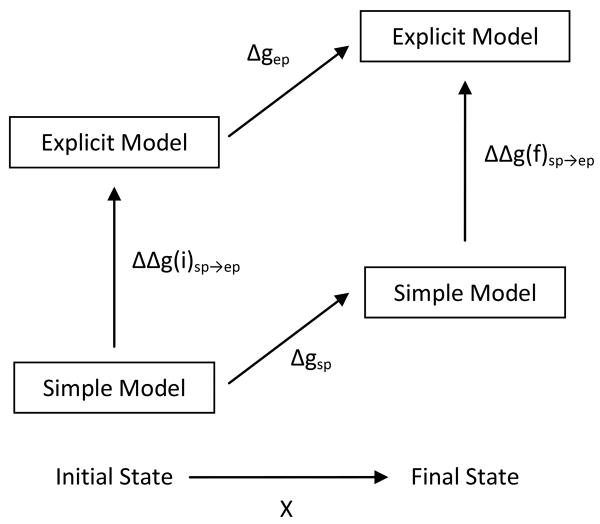
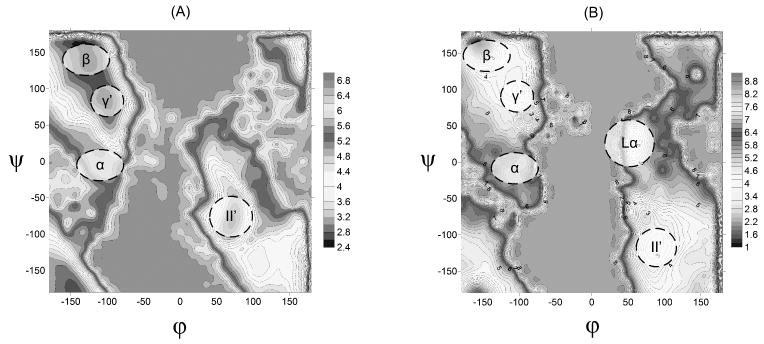
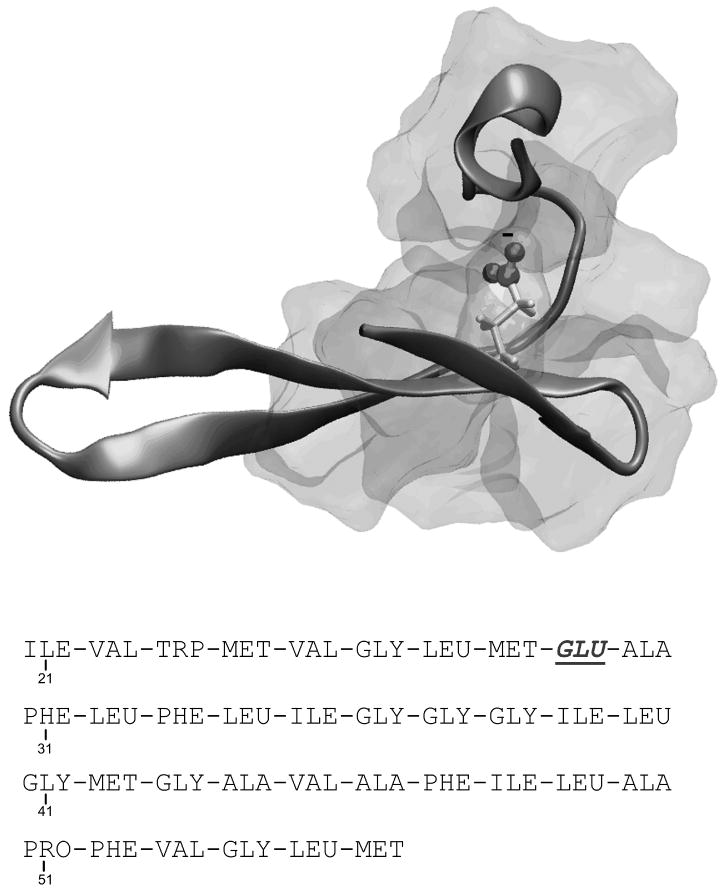
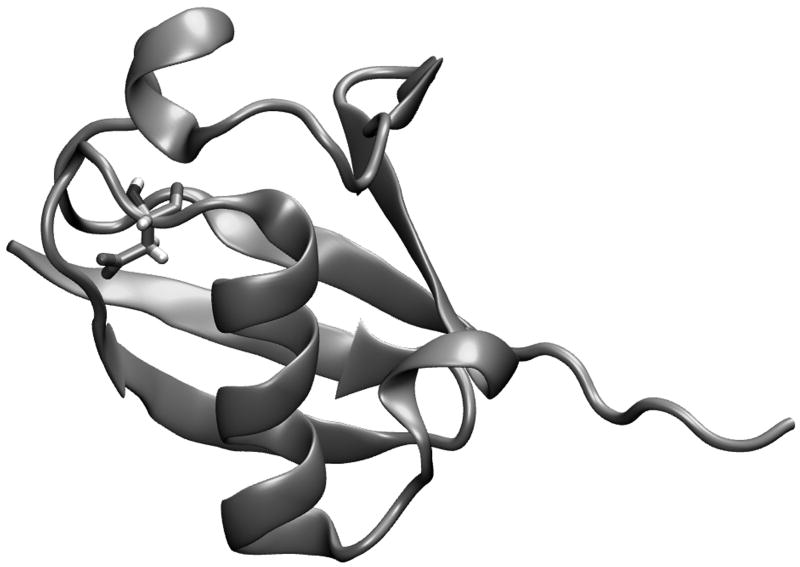
Evaluating the free-energy landscape of proteins and the corresponding functional aspects presents a major challenge for computer simulation approaches. This challenge is due to the complexity of the landscape and the enormous computer time needed for converging simulations. The use of simplified coarse-grained (CG) folding models offers an effective way of sampling the landscape but such a treatment, however, may not give the correct description of the effect of the actual protein residues. A general way around this problem that has been put forward in our early work (Fan et al., Theor Chem Acc 1999;103:77-80) uses the CG model as a reference potential for free-energy calculations of different properties of the explicit model. This method is refined and extended here, focusing on improving the electrostatic treatment and on demonstrating key applications. These applications include: evaluation of changes of folding energy upon mutations, calculations of transition-states binding free energies (which are crucial for rational enzyme design), evaluations of catalytic landscape, and evaluations of the time-dependent responses to pH changes. Furthermore, the general potential of our approach in overcoming major challenges in studies of structure function correlation in proteins is discussed.
Keywords: Coarse Grained model; dielectric constants; free energy calculations; proton transfer.
Figures



References
-
- Levitt M, Warshel A. Computer-Simulation of Protein Folding. Nature. 1975;253(5494):694–698. - PubMed
-
- Dill KA. Dominant Forces in Protein Folding. Biochemistry. 1990;29(31):7133–7155. - PubMed
-
- Olszewski KA, Kolinski A, Skolnick J. Folding simulations and computer redesign of protein A three-helix bundle motifs. Proteins. 1996;25(3):286–299. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
