

Myofilament length dependent activation
- PMID: 20053351
- PMCID: PMC2854194
- DOI: 10.1016/j.yjmcc.2009.12.017
Myofilament length dependent activation
Abstract
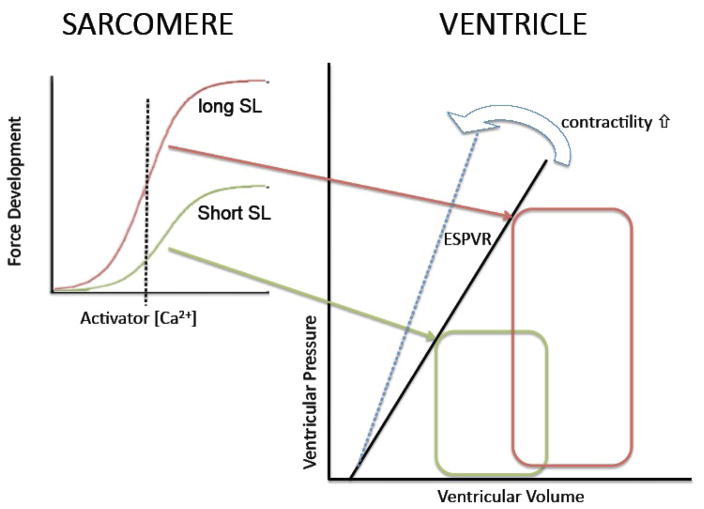
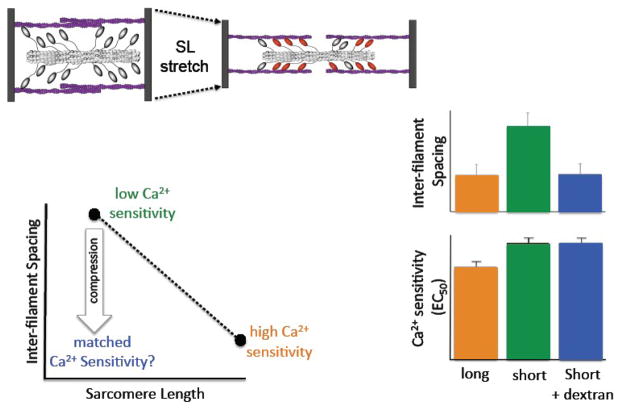
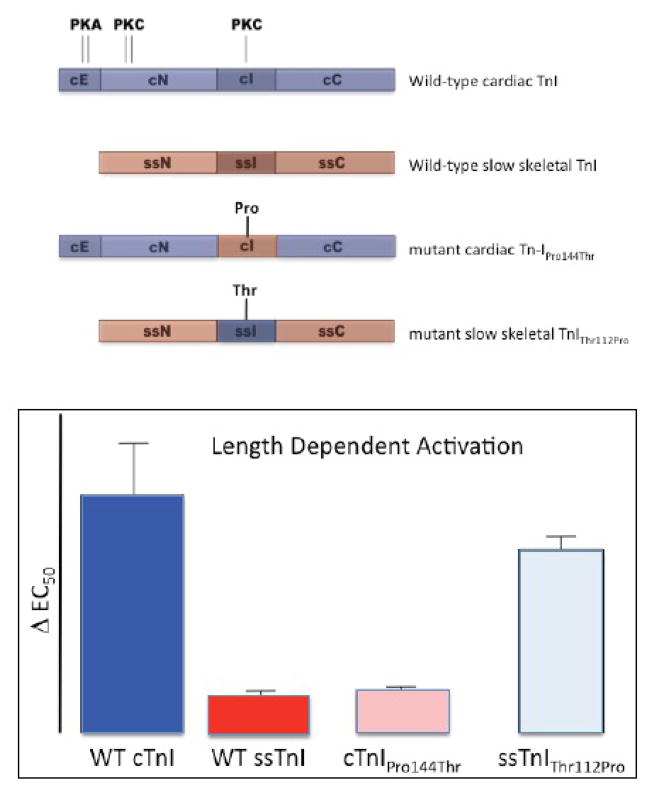
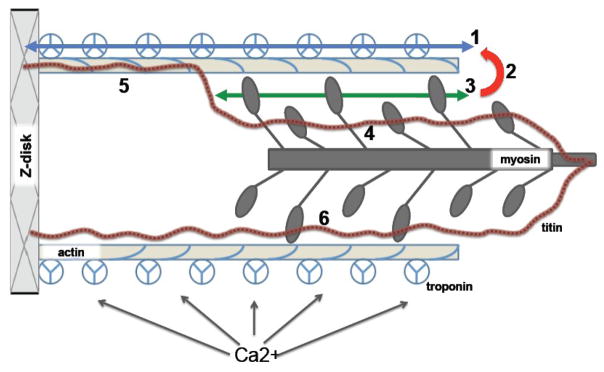
The Frank-Starling law of the heart describes the interrelationship between end-diastolic volume and cardiac ejection volume, a regulatory system that operates on a beat-to-beat basis. The main cellular mechanism that underlies this phenomenon is an increase in the responsiveness of cardiac myofilaments to activating Ca(2+) ions at a longer sarcomere length, commonly referred to as myofilament length-dependent activation. This review focuses on what molecular mechanisms may underlie myofilament length dependency. Specifically, the roles of inter-filament spacing, thick and thin filament based regulation, as well as sarcomeric regulatory proteins are discussed. Although the "Frank-Starling law of the heart" constitutes a fundamental cardiac property that has been appreciated for well over a century, it is still not known in muscle how the contractile apparatus transduces the information concerning sarcomere length to modulate ventricular pressure development.
Copyright (c) 2009 Elsevier Ltd. All rights reserved.
Figures
Comment in
-
Frank-Starling law and mass action calcium activation of the myofibril ATPase; comment on "de Tombe PP, Mateja RD, Tachampa K, Mou YA, Farman GP, Irving TC. Myofilament length dependent activation. J Mol Cell Cardiol 2010; 48: 851-8".J Mol Cell Cardiol. 2010 Oct;49(4):707-8; author reply 709. doi: 10.1016/j.yjmcc.2010.07.003. Epub 2010 Jul 17. J Mol Cell Cardiol. 2010. PMID: 20624395 No abstract available.
References
-
- Sagawa K, Maughan L, Suga H, Sunagawa K. Cardiac contraction and the pressure-volume relationship. New York, Oxford: Oxford University Press; 1988.
-
- de Tombe PP. Cardiac myofilaments: mechanics and regulation. J Biomech. 2003 May;36(5):721–30. - PubMed
-
- Solaro RJ. Integration of myofilament response to Ca2+ with cardiac pump regulation and pump dynamics. Am J Physiol. 1999 Dec;277(6 Pt 2):S155–63. - PubMed
-
- ter Keurs HEDJ, Tyberg JV. Mechanics of the Circulation. Dordrecht: Martinus Nijhoff; 1986.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
