

Sunitinib induces apoptosis and growth arrest of medulloblastoma tumor cells by inhibiting STAT3 and AKT signaling pathways
- PMID: 20053726
- PMCID: PMC2808420
- DOI: 10.1158/1541-7786.MCR-09-0220
Sunitinib induces apoptosis and growth arrest of medulloblastoma tumor cells by inhibiting STAT3 and AKT signaling pathways
Abstract
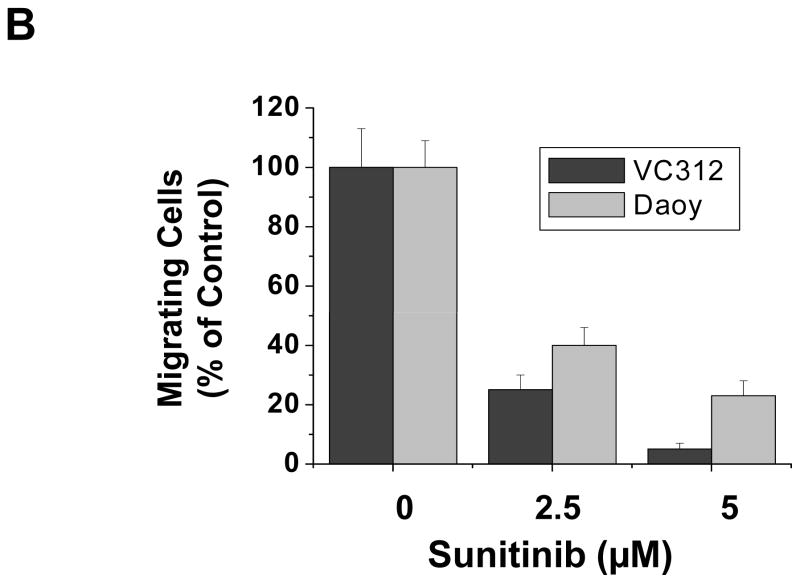
Medulloblastomas are the most frequent malignant brain tumors in children. Sunitinib is an oral multitargeted tyrosine kinase inhibitor used in clinical trials as an antiangiogenic agent for cancer therapy. In this report, we show that sunitinib induced apoptosis and inhibited cell proliferation of both a short-term primary culture (VC312) and an established cell line (Daoy) of human medulloblastomas. Sunitinib treatment resulted in the activation of caspase-3 and cleavage of poly(ADP-ribose) polymerase and upregulation of proapoptotic genes, Bak and Bim, and inhibited the expression of survivin, an antiapoptotic protein. Sunitinib treatment also downregulated cyclin E, cyclin D2, and cyclin D3 and upregulated p21Cip1, all of which are involved in regulating cell cycle. In addition, it inhibited phosphorylation of signal transducer and activator of transcription 3 (STAT3) and AKT (protein kinase B) in the tumor cells. Dephosphorylation of STAT3 (Tyr(705)) induced by sunitinib was helped by a reduction in activities of Janus-activated kinase 2 and Src. Additionally, sodium vanadate, an inhibitor of protein tyrosine phosphatases, partially blocked the inhibition of phosphorylated STAT3 by sunitinib. Loss of phosphorylated AKT after sunitinib treatment was accompanied by decreased phosphorylation of downstream proteins glycogen synthase kinase-3beta and mammalian target of rapamycin. Expression of a constitutively activated STAT3 mutant or myristoylated AKT partially blocked the effects of sunitinib in these tumor cells. Sunitinib also inhibited the migration of medulloblastoma tumor cells in vitro. These findings suggest the potential use of sunitinib for the treatment of pediatric medulloblastomas.
Conflict of interest statement
No potential conflicts of interest were disclosed.
Figures













References
-
- McNeil DE, Cote TR, Clegg L, Rorke LB. Incidence and trends in pediatric malignancies medulloblastoma/primitive neuroectodermal tumor: a SEER update. Surveillance Epidemiology and End Results. Med Pediatr Oncol. 2002;39:190–194. - PubMed
-
- Zurawel RH, Chiappa SA, Allen C, Raffel C. Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res. 1998;58:896–899. - PubMed
-
- Hartmann W, Digon-Sontgerath B, Koch A, Waha A, Endl E, Dani I, et al. Phosphatidylinositol 3′-kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin Cancer Res. 2006;12:3019–3027. - PubMed
-
- Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
