

CREB trans-activation of disruptor of telomeric silencing-1 mediates forskolin inhibition of CTGF transcription in mesangial cells
- PMID: 20053791
- PMCID: PMC2838603
- DOI: 10.1152/ajprenal.00636.2009
CREB trans-activation of disruptor of telomeric silencing-1 mediates forskolin inhibition of CTGF transcription in mesangial cells
Abstract
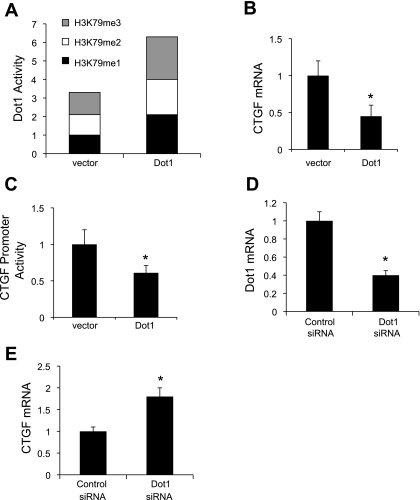
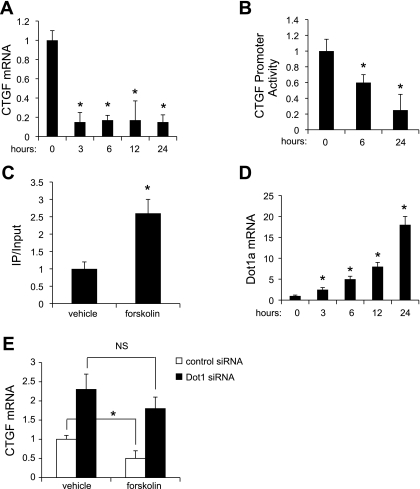
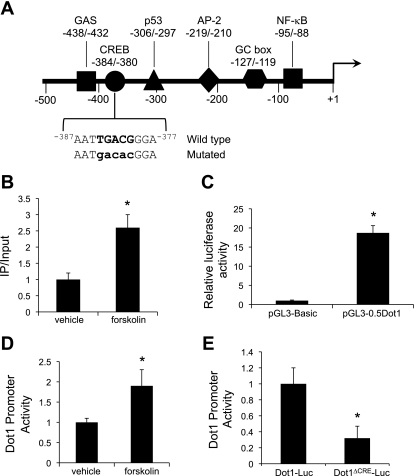
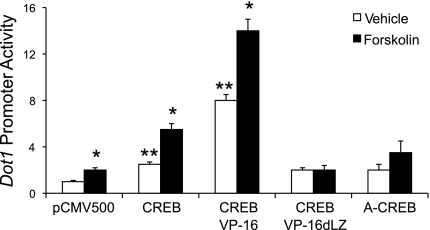
Connective tissue growth factor (CTGF) participates in diverse fibrotic processes including glomerulosclerosis. The adenylyl cyclase agonist forskolin inhibits CTGF expression in mesangial cells by unclear mechanisms. We recently reported that the histone H3K79 methyltransferase disruptor of telomeric silencing-1 (Dot1) suppresses CTGF gene expression in collecting duct cells (J Clin Invest 117: 773-783, 2007) and HEK 293 cells (J Biol Chem In press). In the present study, we characterized the involvement of Dot1 in mediating the inhibitory effect of forskolin on CTGF transcription in mouse mesangial cells. Overexpression of Dot1 or treatment with forskolin dramatically suppressed basal CTGF mRNA levels and CTGF promoter-luciferase activity, while hypermethylating H3K79 in chromatin associated with the CTGF promoter. siRNA knockdown of Dot1 abrogated the inhibitory effect of forskolin on CTGF mRNA expression. Analysis of the Dot1 promoter sequence identified a CREB response element (CRE) at -384/-380. Overexpression of CREB enhanced forskolin-stimulated Dot1 promoter activity. A constitutively active CREB mutant (CREB-VP16) strongly induced Dot1 promoter-luciferase activity, whereas overexpression of CREBdLZ-VP16, which lacks the CREB DNA-binding domain, abolished this activation. Mutation of the -384/-380 CRE resulted in 70% lower levels of Dot1 promoter activity. ChIP assays confirmed CREB binding to the Dot1 promoter in chromatin. We conclude that forskolin stimulates CREB-mediated trans-activation of the Dot1 gene, which leads to hypermethylation of histone H3K79 at the CTGF promoter, and inhibition of CTGF transcription. These data are the first to describe regulation of the Dot1 gene, and disclose a complex network of genetic and epigenetic controls on CTGF transcription.
Figures
Similar articles
-
Sirtuin 1 functionally and physically interacts with disruptor of telomeric silencing-1 to regulate alpha-ENaC transcription in collecting duct.J Biol Chem. 2009 Jul 31;284(31):20917-26. doi: 10.1074/jbc.M109.020073. Epub 2009 Jun 2. J Biol Chem. 2009. PMID: 19491102 Free PMC article.
-
CREB trans-activates the murine H(+)-K(+)-ATPase alpha(2)-subunit gene.Am J Physiol Cell Physiol. 2004 Oct;287(4):C903-11. doi: 10.1152/ajpcell.00065.2004. Epub 2004 May 26. Am J Physiol Cell Physiol. 2004. PMID: 15163620
-
Upregulation of AKT1 protein expression in forskolin-stimulated macrophage: evidence from ChIP analysis that CREB binds to and activates the AKT1 promoter.J Cell Biochem. 2007 Mar 1;100(4):1022-33. doi: 10.1002/jcb.21086. J Cell Biochem. 2007. PMID: 17152074
-
The diverse functions of Dot1 and H3K79 methylation.Genes Dev. 2011 Jul 1;25(13):1345-58. doi: 10.1101/gad.2057811. Genes Dev. 2011. PMID: 21724828 Free PMC article. Review.
-
The histone methyltransferase Dot1/DOT1L as a critical regulator of the cell cycle.Cell Cycle. 2014;13(5):726-38. doi: 10.4161/cc.28104. Epub 2014 Feb 6. Cell Cycle. 2014. PMID: 24526115 Free PMC article. Review.
Cited by
-
Connective tissue growth factor is a target of notch signaling in cells of the osteoblastic lineage.Bone. 2014 Jul;64:273-80. doi: 10.1016/j.bone.2014.04.028. Epub 2014 May 2. Bone. 2014. PMID: 24792956 Free PMC article.
-
Epigenetic Modifications in Essential Hypertension.Int J Mol Sci. 2016 Mar 25;17(4):451. doi: 10.3390/ijms17040451. Int J Mol Sci. 2016. PMID: 27023534 Free PMC article. Review.
-
Cyclic nucleotide signalling in kidney fibrosis.Int J Mol Sci. 2015 Jan 22;16(2):2320-51. doi: 10.3390/ijms16022320. Int J Mol Sci. 2015. PMID: 25622251 Free PMC article. Review.
-
A novel disrupter of telomere silencing 1-like (DOT1L) interaction is required for signal transducer and activator of transcription 1 (STAT1)-activated gene expression.J Biol Chem. 2011 Dec 2;286(48):41195-41204. doi: 10.1074/jbc.M111.284190. Epub 2011 Oct 15. J Biol Chem. 2011. PMID: 22002246 Free PMC article.
-
Gestational age-related patterns of AMOT methylation are revealed in preterm infant endothelial progenitors.PLoS One. 2017 Oct 16;12(10):e0186321. doi: 10.1371/journal.pone.0186321. eCollection 2017. PLoS One. 2017. PMID: 29036193 Free PMC article.
References
-
- Black SA, Jr, Palamakumbura AH, Stan M, Trackman PC. Tissue-specific mechanisms for CCN2/CTGF persistence in fibrotic gingiva: interactions between cAMP and MAPK signaling pathways, and prostaglandin E2-EP3 receptor mediated activation of the c-JUN N-terminal kinase. J Biol Chem 282: 15416–15429, 2007 - PMC - PubMed
-
- Blom IE, van Dijk AJ, Wieten L, Duran K, Ito Y, Kleij L, deNichilo M, Rabelink TJ, Weening JJ, Aten J, Goldschmeding R. In vitro evidence for differential involvement of CTGF, TGFbeta, and PDGF-BB in mesangial response to injury. Nephrol Dial Transplant 16: 1139–1148, 2001 - PubMed
-
- Burns WC, Kantharidis P, Thomas MC. The role of tubular epithelial-mesenchymal transition in progressive kidney disease. Cells Tissues Organs 185: 222–231, 2007 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous