

SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking
- PMID: 20053906
- PMCID: PMC3397691
- DOI: 10.1523/JNEUROSCI.4933-08.2010
SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking
Abstract
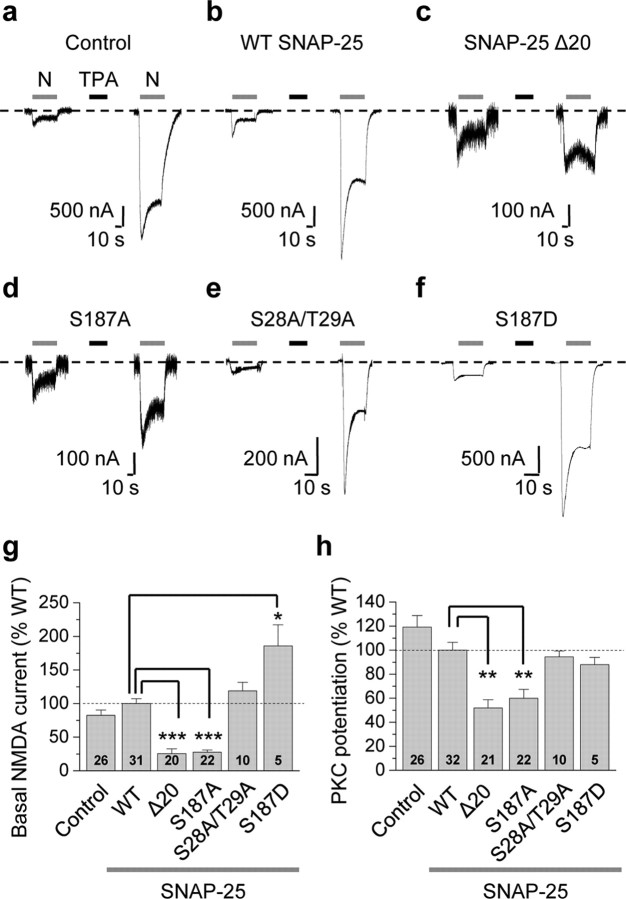
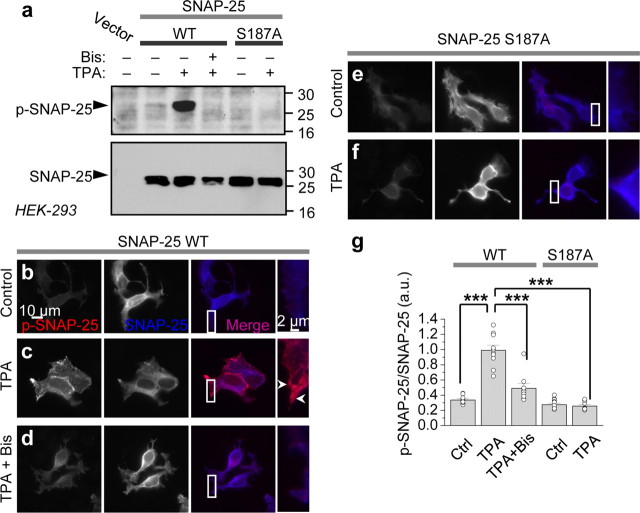
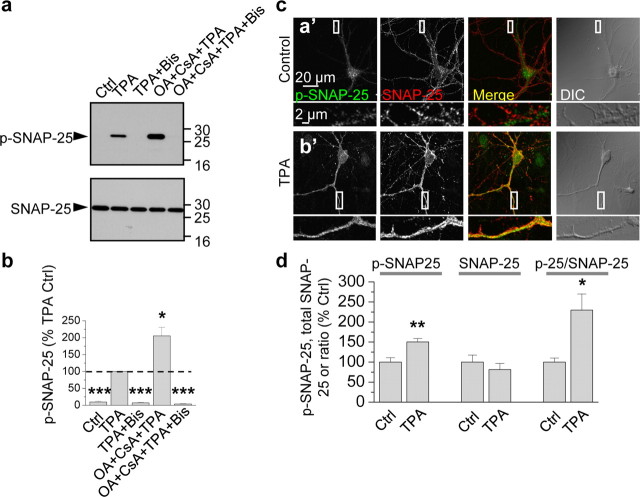
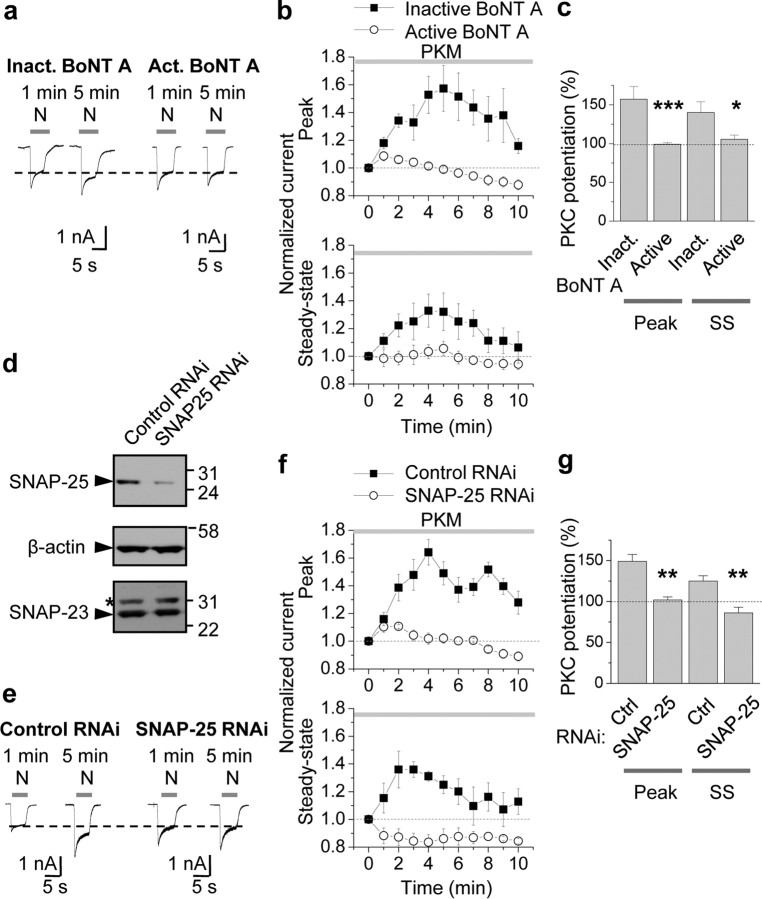
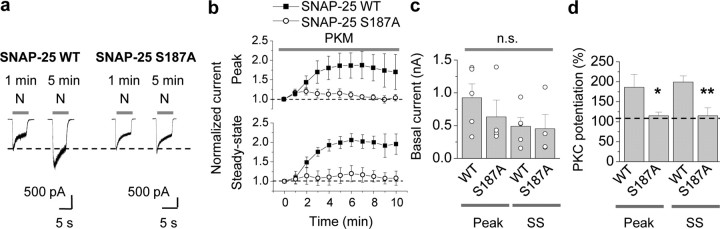
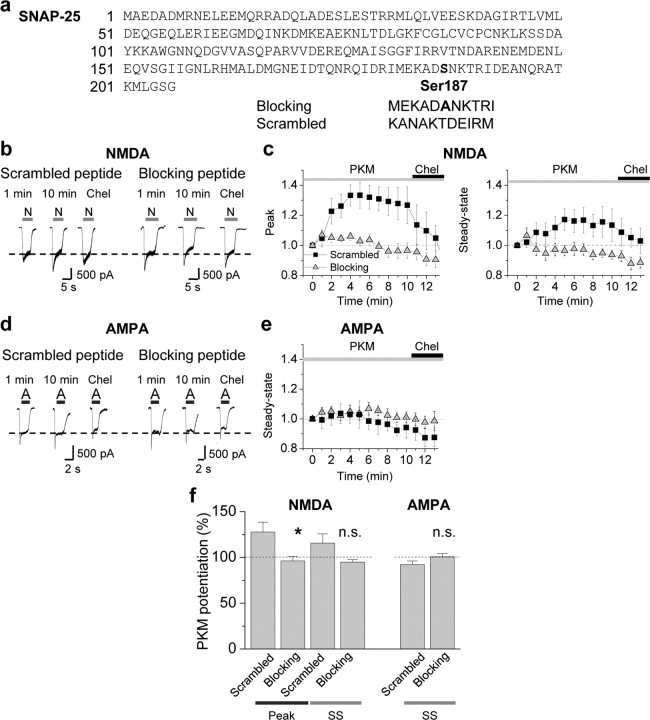
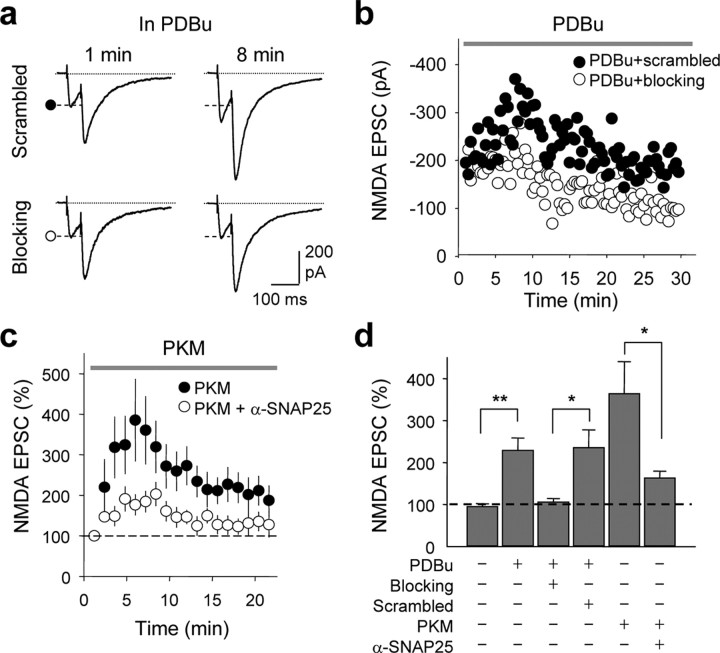
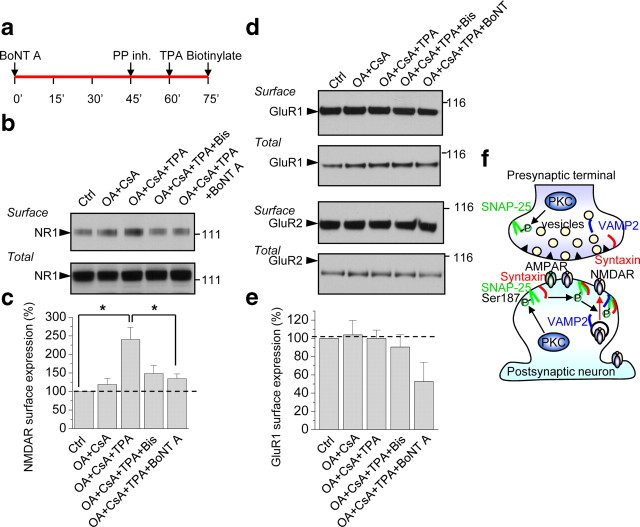
Protein kinase C (PKC) enhances NMDA receptor (NMDAR)-mediated currents and promotes NMDAR delivery to the cell surface via SNARE-dependent exocytosis. Although the mechanisms of PKC potentiation are established, the molecular target of PKC is unclear. Here we show that synaptosomal-associated protein of 25 kDa (SNAP-25), a SNARE protein, is functionally relevant to PKC-dependent NMDAR insertion, and identify serine residue-187 as the molecular target of PKC phosphorylation. Constitutively active PKC delivered via the patch pipette potentiated NMDA (but not AMPA) whole-cell currents in hippocampal neurons. Expression of RNAi targeting SNAP-25 or mutant SNAP-25(S187A) and/or acute disruption of the SNARE complex by treatment with BoNT A, BoNT B or SNAP-25 C-terminal blocking peptide abolished NMDAR potentiation. A SNAP-25 peptide and function-blocking antibody suppressed PKC potentiation of NMDA EPSCs at mossy fiber-CA3 synapses. These findings identify SNAP-25 as the target of PKC phosphorylation critical to PKC-dependent incorporation of synaptic NMDARs and document a postsynaptic action of this major SNARE protein relevant to synaptic plasticity.
Figures
Similar articles
-
Protein kinase C promotes N-methyl-D-aspartate (NMDA) receptor trafficking by indirectly triggering calcium/calmodulin-dependent protein kinase II (CaMKII) autophosphorylation.J Biol Chem. 2011 Jul 15;286(28):25187-200. doi: 10.1074/jbc.M110.192708. Epub 2011 May 23. J Biol Chem. 2011. PMID: 21606495 Free PMC article.
-
Protein kinase C modulates NMDA receptor trafficking and gating.Nat Neurosci. 2001 Apr;4(4):382-90. doi: 10.1038/86028. Nat Neurosci. 2001. PMID: 11276228
-
Protein Kinase C-Mediated Phosphorylation and α2δ-1 Interdependently Regulate NMDA Receptor Trafficking and Activity.J Neurosci. 2021 Jul 28;41(30):6415-6429. doi: 10.1523/JNEUROSCI.0757-21.2021. Epub 2021 Jun 17. J Neurosci. 2021. PMID: 34252035 Free PMC article.
-
Convergence of PKC-dependent kinase signal cascades on NMDA receptors.Curr Drug Targets. 2001 Sep;2(3):299-312. doi: 10.2174/1389450013348452. Curr Drug Targets. 2001. PMID: 11554554 Review.
-
Regulation of exocytosis by protein kinase C.Biochem Soc Trans. 2005 Dec;33(Pt 6):1341-4. doi: 10.1042/BST0331341. Biochem Soc Trans. 2005. PMID: 16246114 Review.
Cited by
-
Electrophysiology of Dendritic Spines: Information Processing, Dynamic Compartmentalization, and Synaptic Plasticity.Adv Neurobiol. 2023;34:103-141. doi: 10.1007/978-3-031-36159-3_3. Adv Neurobiol. 2023. PMID: 37962795
-
Molecular Targets for Combined Therapeutic Strategies to Limit Glioblastoma Cell Migration and Invasion.Front Pharmacol. 2020 Mar 27;11:358. doi: 10.3389/fphar.2020.00358. eCollection 2020. Front Pharmacol. 2020. PMID: 32292341 Free PMC article.
-
Protein kinase C promotes N-methyl-D-aspartate (NMDA) receptor trafficking by indirectly triggering calcium/calmodulin-dependent protein kinase II (CaMKII) autophosphorylation.J Biol Chem. 2011 Jul 15;286(28):25187-200. doi: 10.1074/jbc.M110.192708. Epub 2011 May 23. J Biol Chem. 2011. PMID: 21606495 Free PMC article.
-
Phosphorylation by protein kinase Cα regulates RalB small GTPase protein activation, subcellular localization, and effector utilization.J Biol Chem. 2012 Apr 27;287(18):14827-36. doi: 10.1074/jbc.M112.344986. Epub 2012 Mar 5. J Biol Chem. 2012. PMID: 22393054 Free PMC article.
-
SNARE proteins are essential in the potentiation of NMDA receptors by group II metabotropic glutamate receptors.J Physiol. 2013 Aug 15;591(16):3935-47. doi: 10.1113/jphysiol.2013.255075. Epub 2013 Jun 17. J Physiol. 2013. PMID: 23774277 Free PMC article.
References
-
- Banker G, Goslin K. Developments in neuronal cell culture. Nature. 1988;336:185–186. - PubMed
-
- Ben-Ari Y, Aniksztejn L, Bregestovski P. Protein kinase C modulation of NMDA currents: an important link for LTP induction. Trends Neurosci. 1992;15:333–339. - PubMed
-
- Blanpied TA, Scott DB, Ehlers MD. Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron. 2002;36:435–449. - PubMed
-
- Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, Xu P, Wijayawardana SR, Hanfelt J, Nakagawa T, Sheng M, Peng J. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics. 2006;5:1158–1170. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous