

The impact of macrophage insulin resistance on advanced atherosclerotic plaque progression
- PMID: 20056946
- PMCID: PMC2805467
- DOI: 10.1161/CIRCRESAHA.109.208488
The impact of macrophage insulin resistance on advanced atherosclerotic plaque progression
Abstract
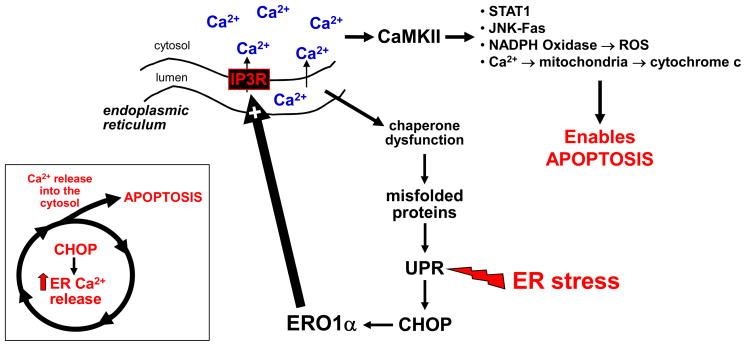
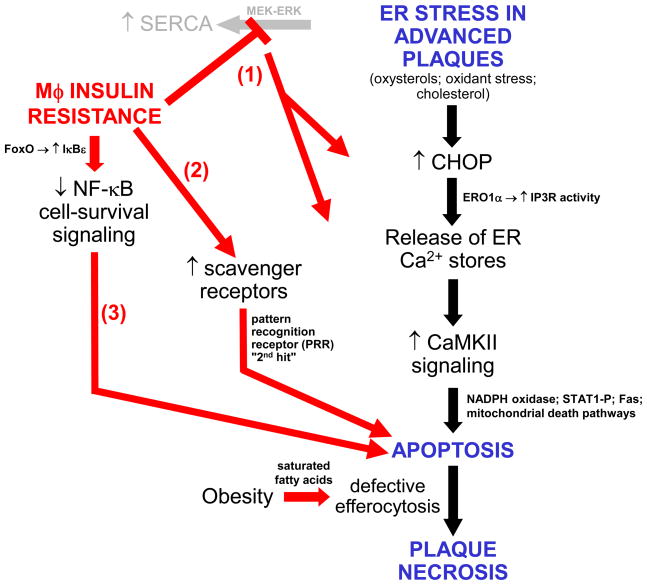
Atherothrombotic vascular disease is the major cause of death and disability in obese and diabetic subjects with insulin resistance. Although increased systemic risk factors in the setting of insulin resistance contribute to this problem, it is likely exacerbated by direct effects of insulin resistance on the arterial wall cells that participate in atherosclerosis. A critical process in the progression of subclinical atherosclerotic lesions to clinically relevant lesions is necrotic breakdown of plaques. Plaque necrosis, which is particularly prominent in the lesions of diabetics, is caused by the combination of macrophage apoptosis and defective phagocytic clearance, or efferocytosis, of the apoptotic macrophages. One cause of macrophage apoptosis in advanced plaques is activation of a proapoptotic branch of the unfolded protein response, which is an endoplasmic reticulum stress pathway. Macrophages have a functional insulin receptor signaling pathway, and downregulation of this pathway in the setting insulin resistance enhances unfolded protein response-induced apoptosis. Moreover, other aspects of the obesity/insulin-resistance syndrome may adversely affect efferocytosis. These processes may therefore provide an important mechanistic link among insulin resistance, plaque necrosis, and atherothrombotic vascular disease and suggest novel therapeutic approaches to this expanding health problem.
Figures
References
-
- Yach D, Stuckler D, Brownell KD. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nat Med. 2006;12:62–66. - PubMed
-
- Fox CS, Coady S, Sorlie PD, D’Agostino RB, Sr, Pencina MJ, Vasan RS, Meigs JB, Levy D, Savage PJ. Increasing cardiovascular disease burden due to diabetes mellitus: the Framingham Heart Study. Circulation. 2007;115:1544–1550. - PubMed
-
- Grundy SM. Obesity, metabolic syndrome, and cardiovascular disease. J Clin Endocrinol Metab. 2004;89:2595–2600. - PubMed
-
- Haffner SM, D’Agostino R, Jr, Mykkanen L, Tracy R, Howard B, Rewers M, Selby J, Savage PJ, Saad MF. Insulin sensitivity in subjects with type 2 diabetes. Relationship to cardiovascular risk factors: the Insulin Resistance Atherosclerosis Study. Diabetes Care. 1999;22:562–568. - PubMed
-
- Gami AS, Witt BJ, Howard DE, Erwin PJ, Gami LA, Somers VK, Montori VM. Metabolic syndrome and risk of incident cardiovascular events and death: a systematic review and meta-analysis of longitudinal studies. J Am Coll Cardiol. 2007;49:403–414. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
