

Pathophysiology of thrombotic thrombocytopenic purpura
- PMID: 20058209
- PMCID: PMC3159000
- DOI: 10.1007/s12185-009-0476-1
Pathophysiology of thrombotic thrombocytopenic purpura
Abstract
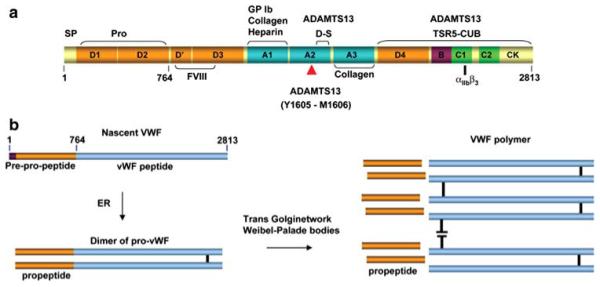
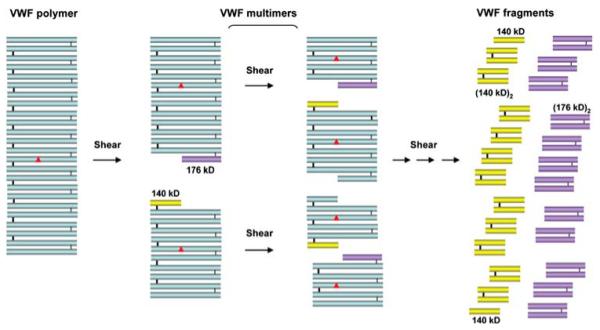
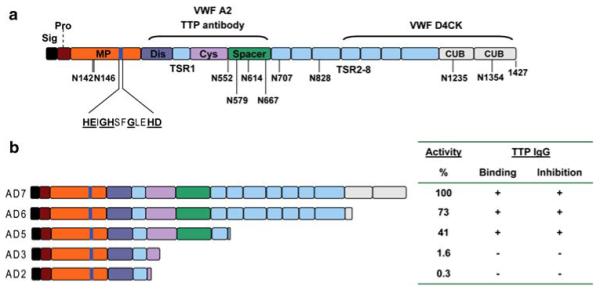
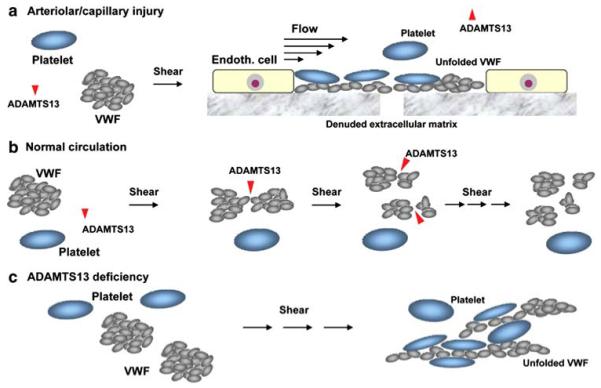
Thrombotic thrombocytopenic purpura (TTP) is a disorder with characteristic von Willebrand factor (VWF)-rich microthrombi affecting the arterioles and capillaries of multiple organs. The disorder frequently leads to early death unless the patients are treated with plasma exchange or infusion. Studies in the last decade have provided ample evidence to support that TTP is caused by deficiency of a plasma metalloprotease, ADAMTS13. When exposed to high shear stress in the microcirculation, VWF and platelets are prone to form aggregates. This propensity of VWF and platelet to form microvascular thrombosis is mitigated by ADAMTS13, which cleaves VWF before it is activated by shear stress to cause platelet aggregation in the circulation. Deficiency of ADAMTS13, due to autoimmune inhibitors in patients with acquired TTP and mutations of the ADAMTS13 gene in hereditary cases, leads to VWF-platelet aggregation and microvascular thrombosis of TTP. In this review, we discuss the current knowledge on the pathogenesis, diagnosis and management of TTP, address the ongoing controversies, and indicate the directions of future investigations.
Figures
References
-
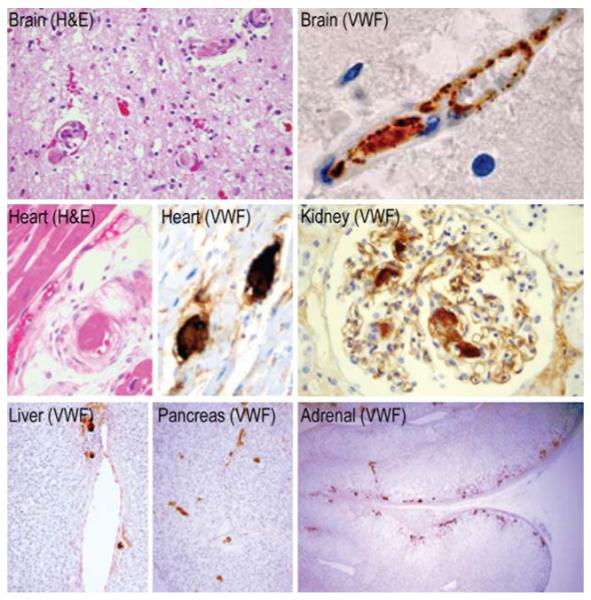
- Asada Y, Sumiyoshi A, Hayashi T, Suzumiya J, Kaketani K. Immunohistochemistry of vascular lesion in thrombotic thrombocytopenic purpura, with special reference to factor VIII related antigen. Thromb Res. 1985;38:469–79. - PubMed
-
- Tsai HM, Chandler WL, Sarode R, et al. von Willebrand factor and von Willebrand factor-cleaving metalloprotease activity in Escherichia coli O157:H7-associated hemolytic uremic syndrome. Pediatr Res. 2001;49:653–9. - PubMed
-
- Hosler GA, Cusumano AM, Hutchins GM. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are distinct pathologic entities. A review of 56 autopsy cases. Arch Pathol Lab Med. 2003;127:834–9. - PubMed
-
- Sadler JE. New concepts in von Willebrand disease. Annu Rev Med. 2005;56:173–91. - PubMed
-
- Tsai HM, Nagel RL, Hatcher VB, Sussman II. Multimeric composition of endothelial cell-derived von Willebrand factor. Blood. 1989;73:2074–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
