

Role of oxidatively induced DNA lesions in human pathogenesis
- PMID: 20060490
- PMCID: PMC3074954
- DOI: 10.1016/j.mrrev.2009.12.005
Role of oxidatively induced DNA lesions in human pathogenesis
Abstract
Genome stability is essential for maintaining cellular and organismal homeostasis, but it is subject to many threats. One ubiquitous threat is from a class of compounds known as reactive oxygen species (ROS), which can indiscriminately react with many cellular biomolecules including proteins, lipids, and DNA to produce a variety of oxidative lesions. These DNA oxidation products are a direct risk to genome stability, and of particular importance are oxidative clustered DNA lesions (OCDLs), defined as two or more oxidative lesions present within 10 bp of each other. ROS can be produced by exposure of cells to exogenous environmental agents including ionizing radiation, light, chemicals, and metals. In addition, they are produced by cellular metabolism including mitochondrial ATP generation. However, ROS also serve a variety of critical cellular functions and optimal ROS levels are maintained by multiple cellular antioxidant defenses. Oxidative DNA lesions can be efficiently repaired by base excision repair or nucleotide excision repair. If ROS levels increase beyond the capacity of its antioxidant defenses, the cell's DNA repair capacity can become overwhelmed, leading to the accumulation of oxidative DNA damage products including OCDLs, which are more difficult to repair than individual isolated DNA damage products. Here we focus on the induction and repair of OCDLs and other oxidatively induced DNA lesions. If unrepaired, these lesions can lead to the formation of mutations, DNA DSBs, and chromosome abnormalities. We discuss the roles of these lesions in human pathologies including aging and cancer, and in bystander effects.
Published by Elsevier B.V.
Figures
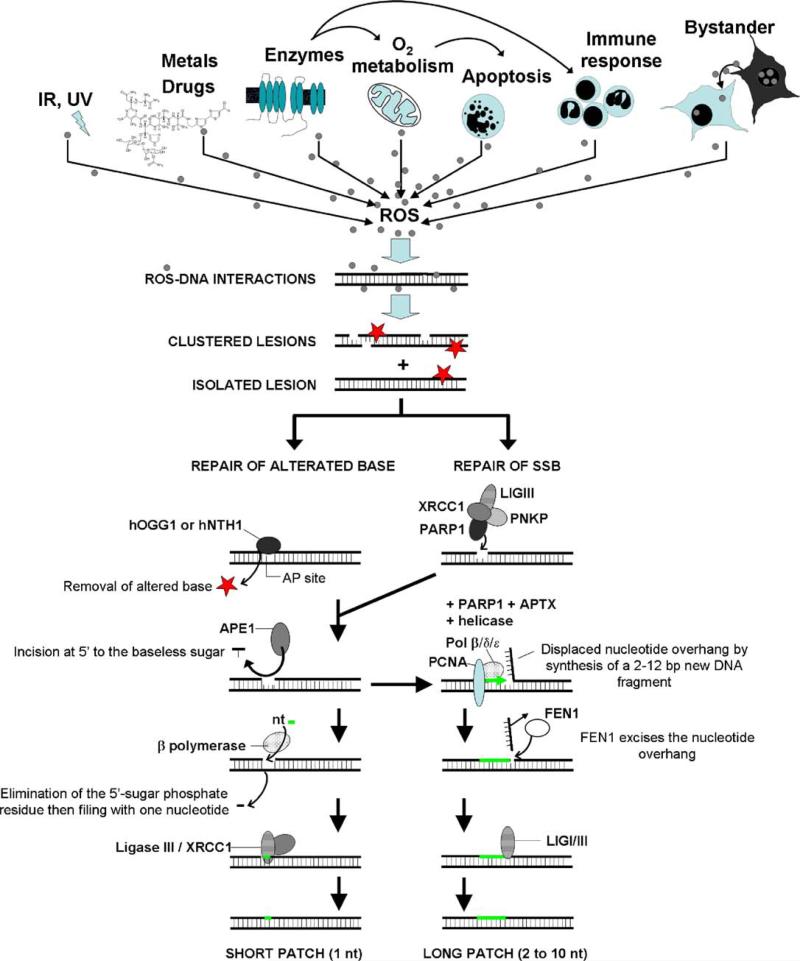
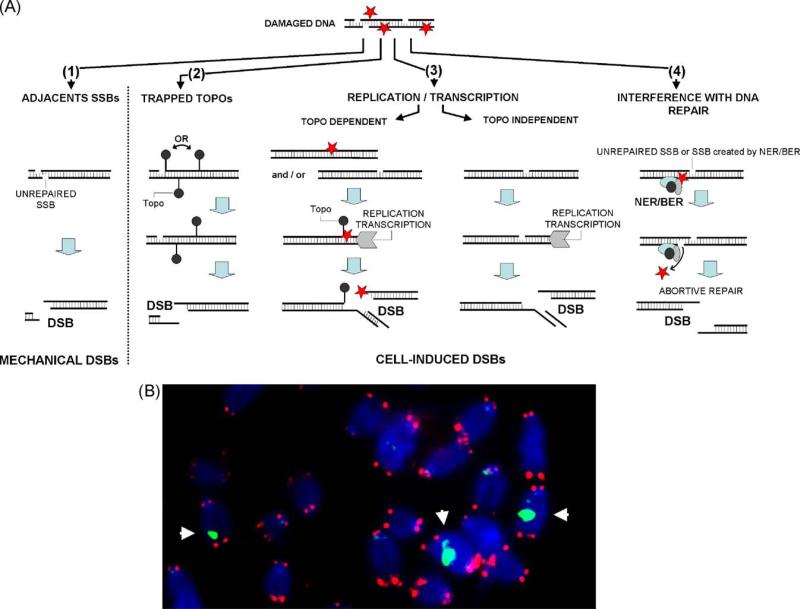
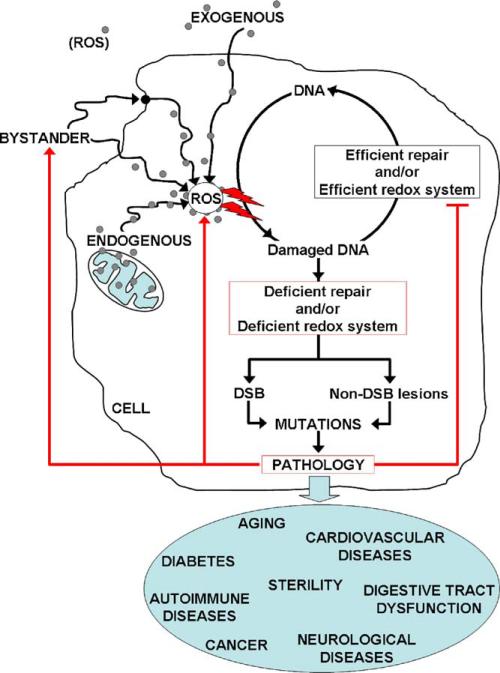
 : increase of ROS production. T: decrease of DNA repair or redox capacity.
: increase of ROS production. T: decrease of DNA repair or redox capacity.References
-
- Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001;31:1287–1312. - PubMed
-
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. - PubMed
-
- Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat. Rev. Cancer. 2003;3:276–285. - PubMed
-
- O'Neill P, Wardman P. Radiation chemistry comes before radiation biology. Int. J. Radiat. Biol. 2009;85:9–25. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
