

Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells
- PMID: 20060889
- PMCID: PMC2861908
- DOI: 10.1016/j.freeradbiomed.2009.12.024
Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells
Abstract
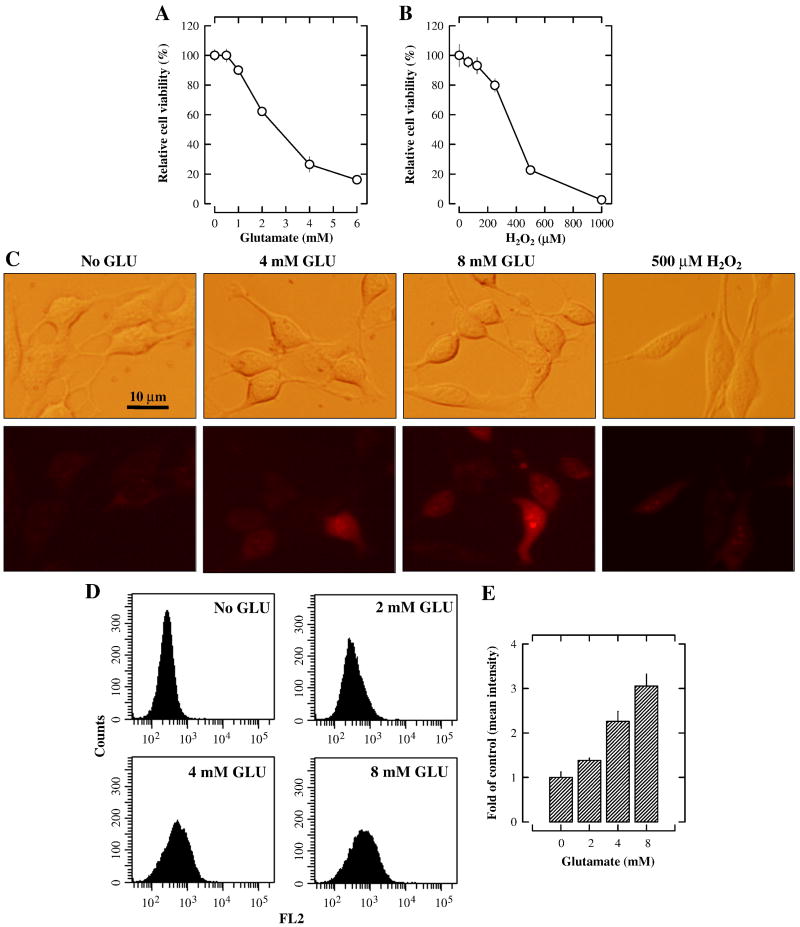
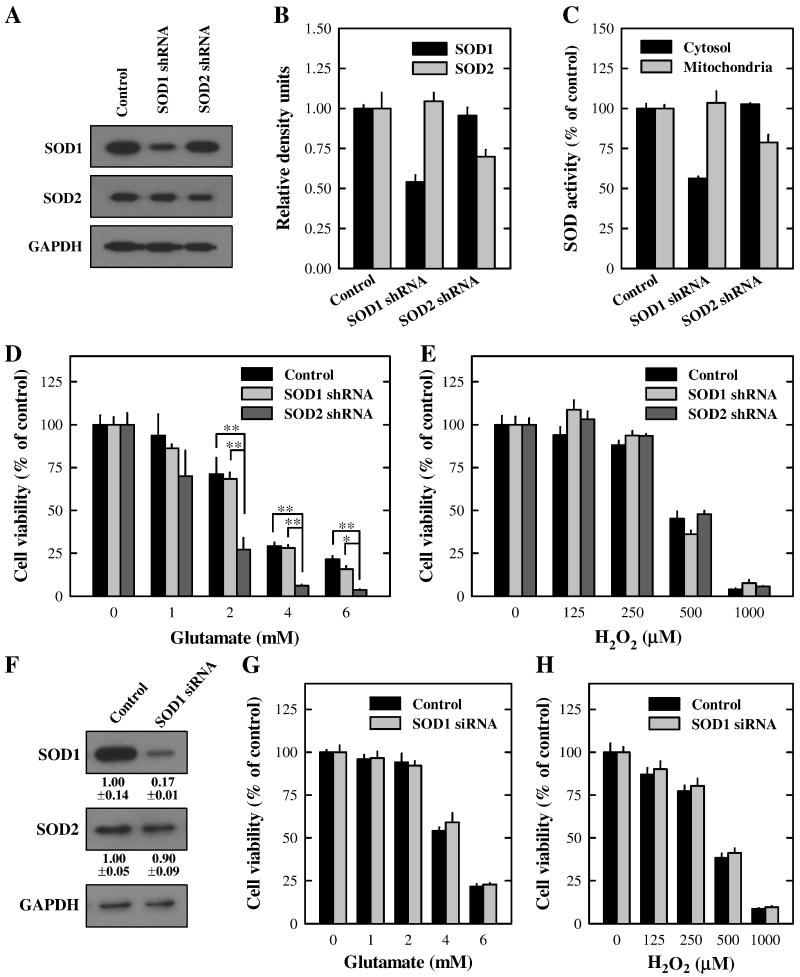
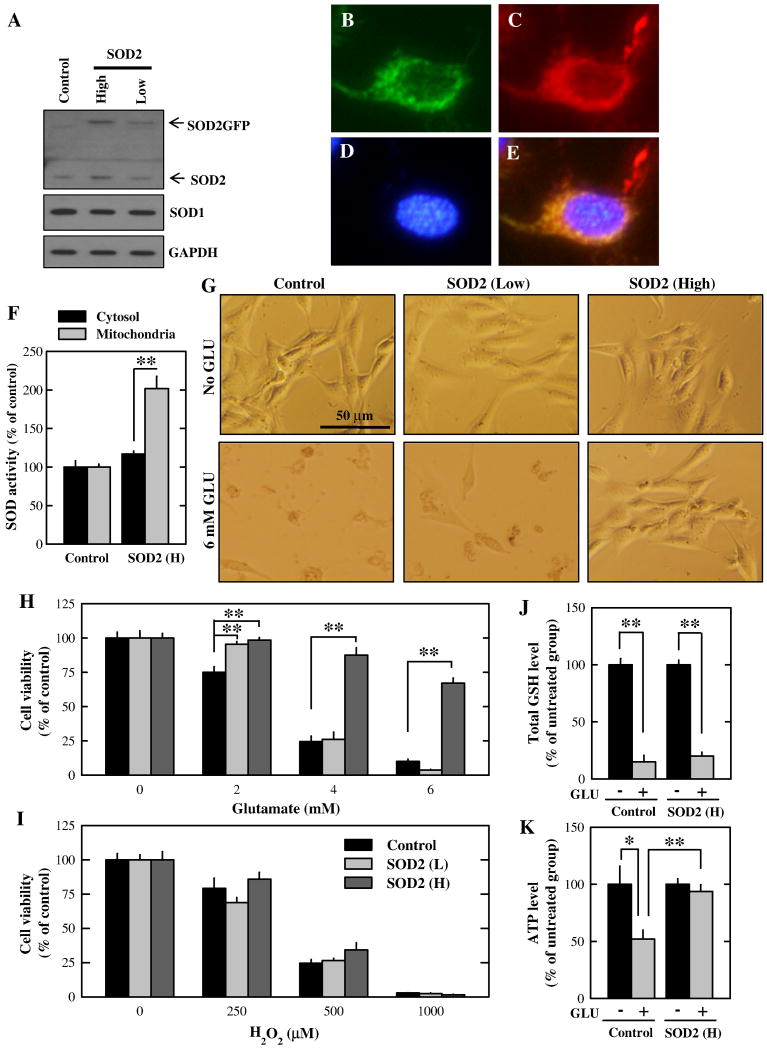
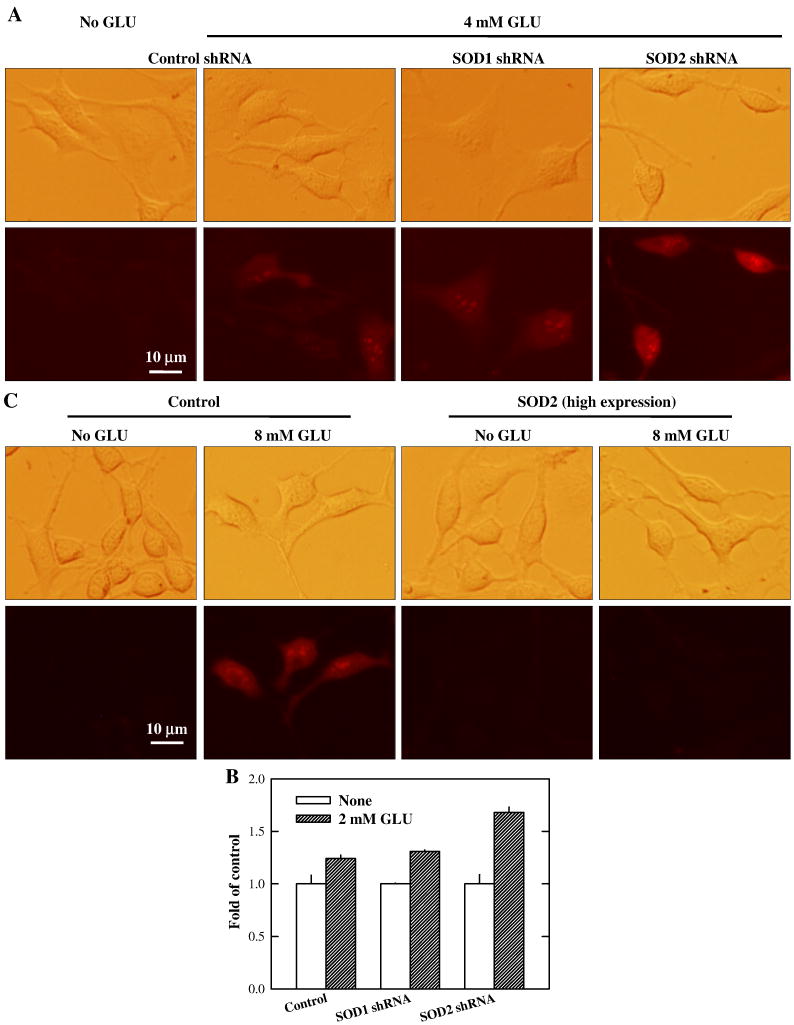
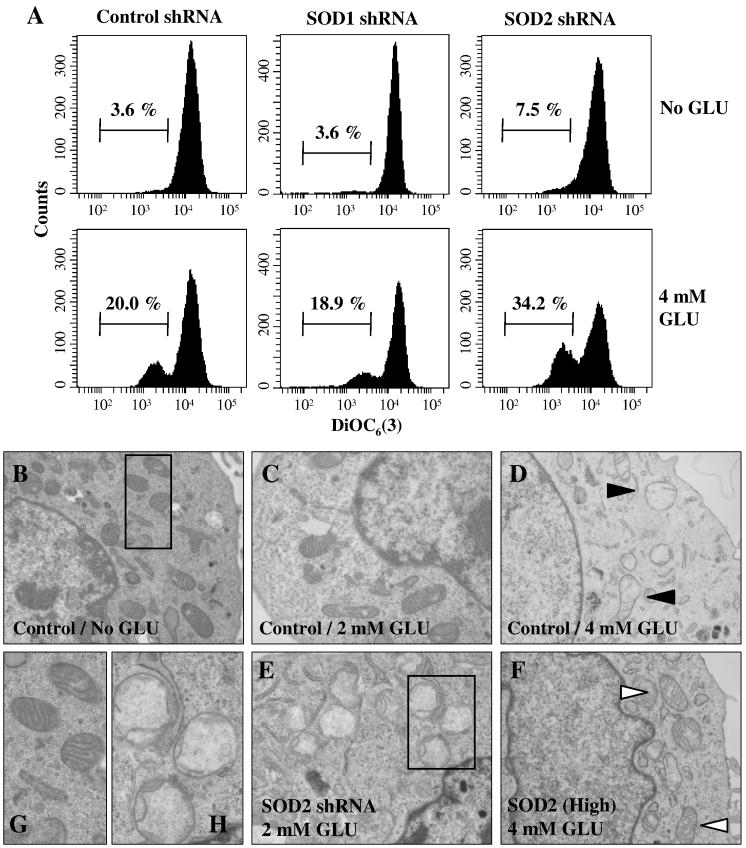
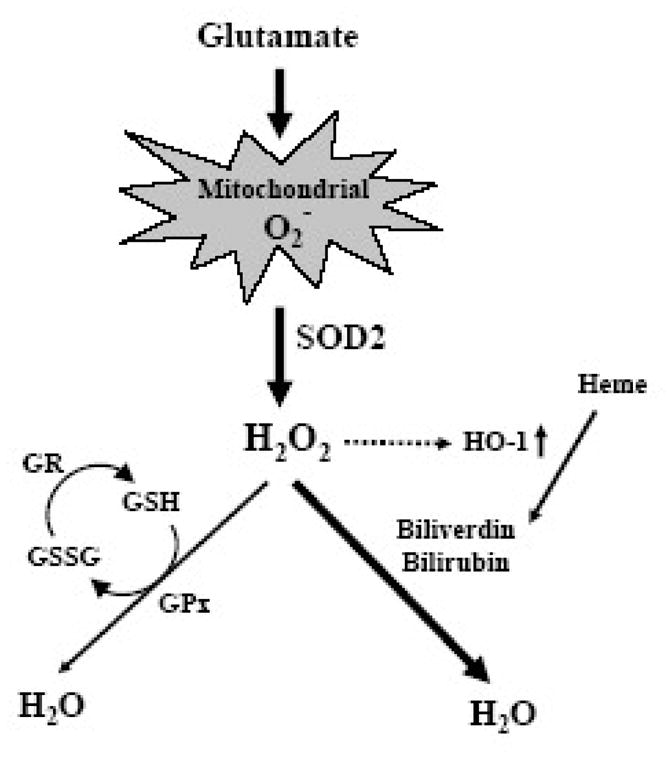
Oxidative cell death is an important contributing factor in neurodegenerative diseases. Using HT22 mouse hippocampal neuronal cells as a model, we sought to demonstrate that mitochondria are crucial early targets of glutamate-induced oxidative cell death. We show that when HT22 cells were transfected with shRNA for knockdown of the mitochondrial superoxide dismutase (SOD2), these cells became more susceptible to glutamate-induced oxidative cell death. The increased susceptibility was accompanied by increased accumulation of mitochondrial superoxide and loss of normal mitochondrial morphology and function at early time points after glutamate exposure. However, overexpression of SOD2 in these cells reduced the mitochondrial superoxide level, protected mitochondrial morphology and functions, and provided resistance against glutamate-induced oxidative cytotoxicity. The change in the sensitivity of these SOD2-altered HT22 cells was neurotoxicant-specific, because the cytotoxicity of hydrogen peroxide was not altered in these cells. In addition, selective knockdown of the cytosolic SOD1 in cultured HT22 cells did not appreciably alter their susceptibility to either glutamate or hydrogen peroxide. These findings show that the mitochondrial SOD2 plays a critical role in protecting neuronal cells from glutamate-induced oxidative stress and cytotoxicity. These data also indicate that mitochondria are important early targets of glutamate-induced oxidative neurotoxicity.
Copyright 2010 Elsevier Inc. All rights reserved.
Figures
References
-
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. - PubMed
-
- Blandini F, Porter RH, Greenamyre JT. Glutamate and Parkinson’s desease. Mol Neurobiol. 1996;12:73–94. - PubMed
-
- Choi DW. Glutamate neurotoxicity and diseases of nervous system. Neuron. 1988;1:623–634. - PubMed
-
- Choi DW. Exitotoxic cell death. J Neurobiol. 1992;23:1261–1276. - PubMed
-
- Tan S, Wood M, Maher P. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J Neurochem. 1998;71:95–105. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
