

Compared effects of missense mutations in Very-Long-Chain Acyl-CoA Dehydrogenase deficiency: Combined analysis by structural, functional and pharmacological approaches
- PMID: 20060901
- PMCID: PMC3401415
- DOI: 10.1016/j.bbadis.2010.01.001
Compared effects of missense mutations in Very-Long-Chain Acyl-CoA Dehydrogenase deficiency: Combined analysis by structural, functional and pharmacological approaches
Abstract
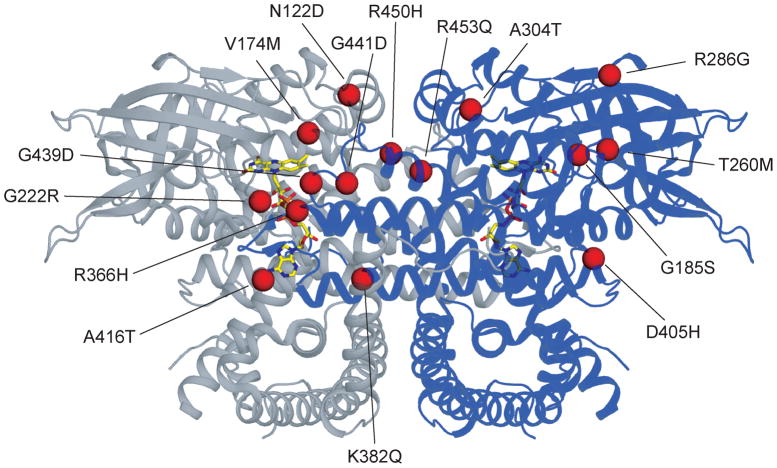
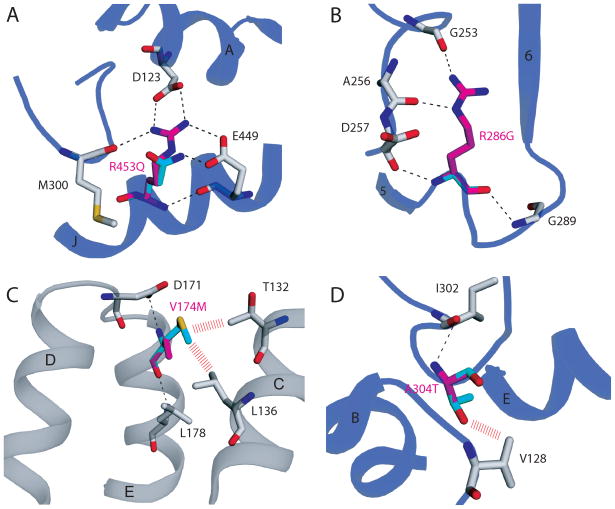
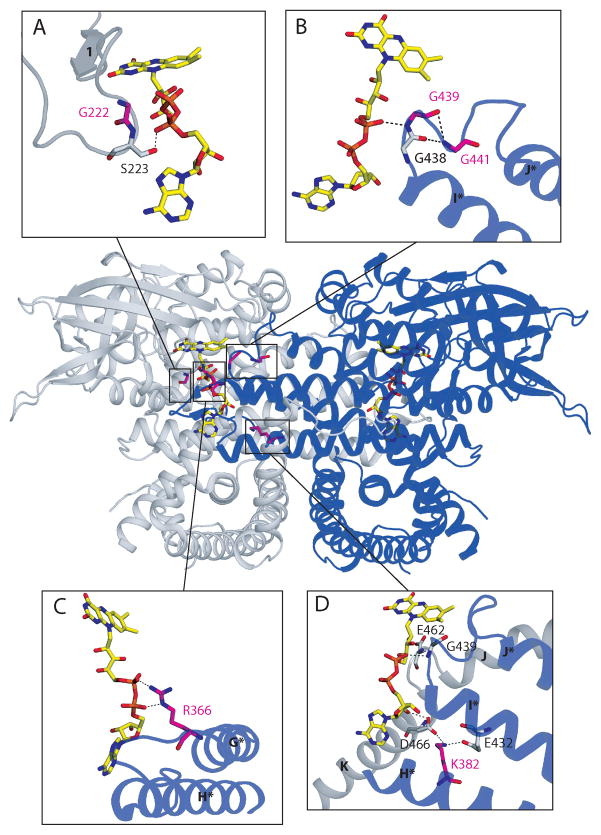
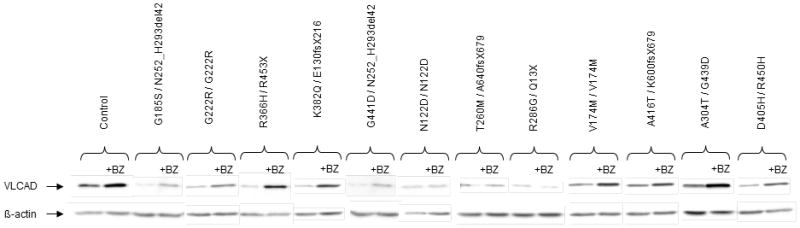
Very-Long-Chain Acyl-CoA Dehydrogenase deficiency (VLCADD) is an autosomal recessive disorder considered as one of the more common ss-oxidation defects, possibly associated with neonatal cardiomyopathy, infantile hepatic coma, or adult-onset myopathy. Numerous gene missense mutations have been described in these VLCADD phenotypes, but only few of them have been structurally and functionally analyzed, and the molecular basis of disease variability is still poorly understood. To address this question, we first analyzed fourteen disease-causing amino acid changes using the recently described crystal structure of VLCAD. The predicted effects varied from the replacement of amino acid residues lining the substrate binding cavity, involved in holoenzyme-FAD interactions or in enzyme dimerisation, predicted to have severe functional consequences, up to amino acid substitutions outside key enzyme domains or lying on near enzyme surface, with predicted milder consequences. These data were combined with functional analysis of residual fatty acid oxidation (FAO) and VLCAD protein levels in patient cells harboring these mutations, before and after pharmacological stimulation by bezafibrate. Mutations identified as detrimental to the protein structure in the 3-D model were generally associated to profound FAO and VLCAD protein deficiencies in the patient cells, however, some mutations affecting FAD binding or monomer-monomer interactions allowed a partial response to bezafibrate. On the other hand, bezafibrate restored near-normal FAO rates in some mutations predicted to have milder consequences on enzyme structure. Overall, combination of structural, biochemical, and pharmacological analysis allowed assessment of the relative severity of individual mutations, with possible applications for disease management and therapeutic approach.
Copyright 2010 Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders.Hum Mol Genet. 2005 Sep 15;14(18):2695-703. doi: 10.1093/hmg/ddi303. Epub 2005 Aug 22. Hum Mol Genet. 2005. PMID: 16115821
-
Genetic basis for correction of very-long-chain acyl-coenzyme A dehydrogenase deficiency by bezafibrate in patient fibroblasts: toward a genotype-based therapy.Am J Hum Genet. 2007 Dec;81(6):1133-43. doi: 10.1086/522375. Epub 2007 Oct 29. Am J Hum Genet. 2007. PMID: 17999356 Free PMC article.
-
Effect of heat stress and bezafibrate on mitochondrial beta-oxidation: comparison between cultured cells from normal and mitochondrial fatty acid oxidation disorder children using in vitro probe acylcarnitine profiling assay.Brain Dev. 2010 May;32(5):362-70. doi: 10.1016/j.braindev.2009.06.001. Epub 2009 Jul 8. Brain Dev. 2010. PMID: 19589653
-
The Pathogenesis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency.Biomolecules. 2025 Mar 14;15(3):416. doi: 10.3390/biom15030416. Biomolecules. 2025. PMID: 40149952 Free PMC article. Review.
-
Genetic and cellular modifiers of oxidative stress: what can we learn from fatty acid oxidation defects?Mol Genet Metab. 2013;110 Suppl:S31-9. doi: 10.1016/j.ymgme.2013.10.007. Epub 2013 Oct 12. Mol Genet Metab. 2013. PMID: 24206932 Review.
Cited by
-
Elucidating the Beneficial Role of PPAR Agonists in Cardiac Diseases.Int J Mol Sci. 2018 Nov 4;19(11):3464. doi: 10.3390/ijms19113464. Int J Mol Sci. 2018. PMID: 30400386 Free PMC article. Review.
-
Disturbance of Mitochondrial Dynamics, Endoplasmic Reticulum-Mitochondria Crosstalk, Redox Homeostasis, and Inflammatory Response in the Brain of Glutaryl-CoA Dehydrogenase-Deficient Mice: Neuroprotective Effects of Bezafibrate.Mol Neurobiol. 2022 Aug;59(8):4839-4853. doi: 10.1007/s12035-022-02887-3. Epub 2022 May 31. Mol Neurobiol. 2022. PMID: 35639256
-
Four novel variants identified in the ACADVL gene causing very-long-chain acyl-coenzyme A dehydrogenase deficiency in four unrelated Chinese families.Front Genet. 2024 Aug 12;15:1433160. doi: 10.3389/fgene.2024.1433160. eCollection 2024. Front Genet. 2024. PMID: 39188284 Free PMC article.
-
Molecular and cellular pathology of very-long-chain acyl-CoA dehydrogenase deficiency.Mol Genet Metab. 2013 May;109(1):21-7. doi: 10.1016/j.ymgme.2013.02.002. Epub 2013 Feb 13. Mol Genet Metab. 2013. PMID: 23480858 Free PMC article.
-
Treatment of VLCAD-Deficient Patient Fibroblasts with Peroxisome Proliferator-Activated Receptor δ Agonist Improves Cellular Bioenergetics.Cells. 2022 Aug 24;11(17):2635. doi: 10.3390/cells11172635. Cells. 2022. PMID: 36078043 Free PMC article.
References
-
- Arnold GL, Van Hove J, Freedenberg D, Strauss A, Longo N, Burton B, Garganta C, Ficicioglu C, Cederbaum S, Harding C, Boles RG, Matern D, Chakraborty P, Feigenbaum A. A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2009;96:85–90. - PMC - PubMed
-
- Andresen BS, Olpin S, Poorthuis BJ, Scholte HR, Vianey-Saban C, Wanders R, Ijlst L, Morris A, Pourfarzam M, Bartlett K, Baumgartner ER, deKlerk JB, Schroeder LD, Corydon TJ, Lund H, Winter V, Bross P, Bolund L, Gregersen N. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet. 1999;64:479–94. - PMC - PubMed
-
- Liebig M, Schymik I, Mueller M, Wendel U, Mayatepek E, Ruiter J, Strauss AW, Wanders RJ, Spiekerkoetter U. Neonatal screening for very long-chain acyl-coA dehydrogenase deficiency: enzymatic and molecular evaluation of neonates with elevated C14:1-carnitine levels. Pediatrics. 2006;118:1065–9. - PubMed
-
- Gregersen N, Andresen BS, Pedersen CB, Olsen RK, Corydon TJ, Bross P. Mitochondrial fatty acid oxidation defects--remaining challenges. J Inherit Metab Dis. 2008;31:643–57. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
