

Classical maple syrup urine disease and brain development: principles of management and formula design
- PMID: 20061171
- PMCID: PMC3671925
- DOI: 10.1016/j.ymgme.2009.12.007
Classical maple syrup urine disease and brain development: principles of management and formula design
Erratum in
- Mol Genet Metab. 2011 Jun;103(2):202. Shelmer, Diana [corrected to Shellmer, Diana]
Abstract
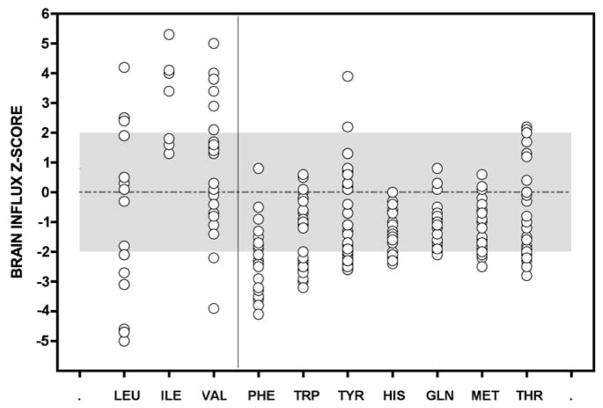
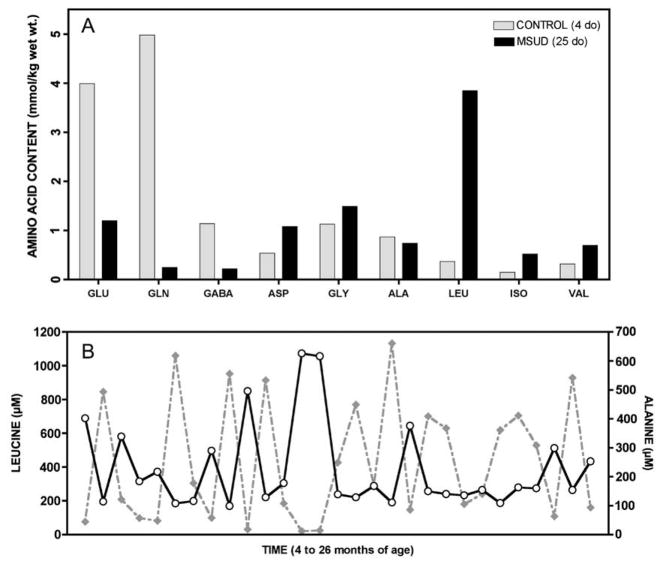
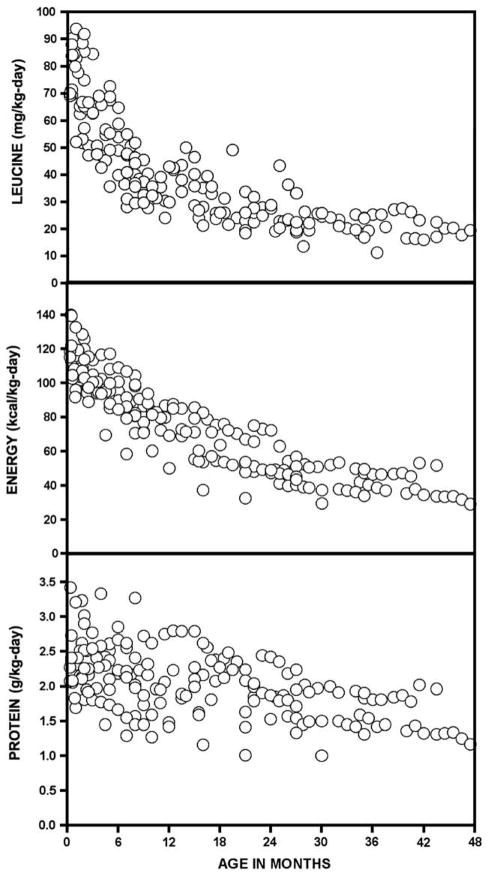
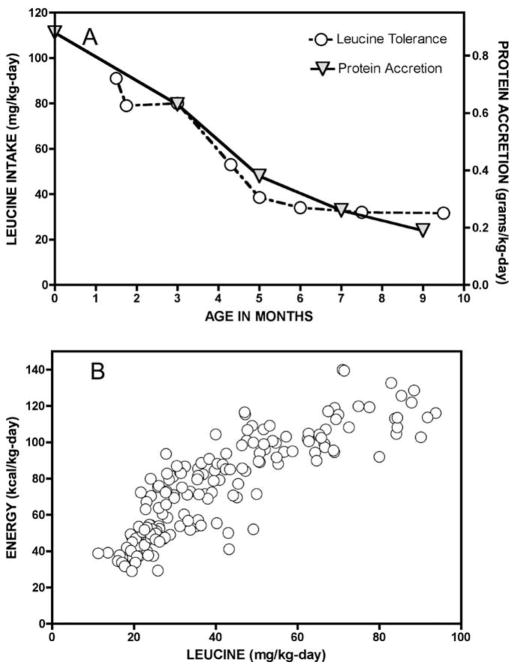
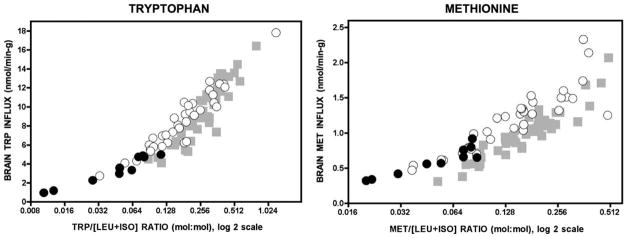
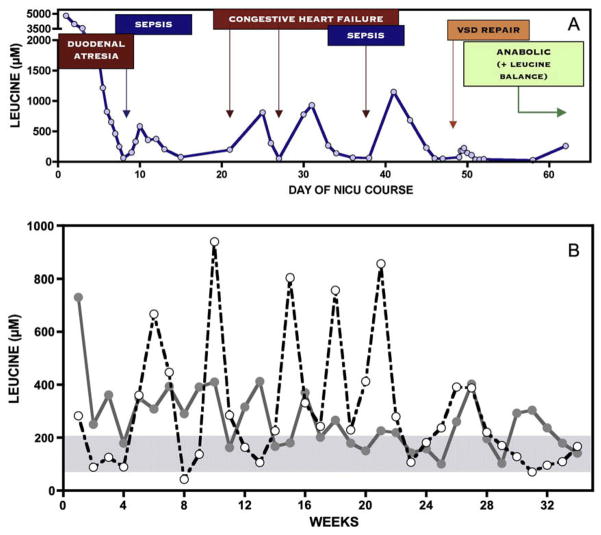
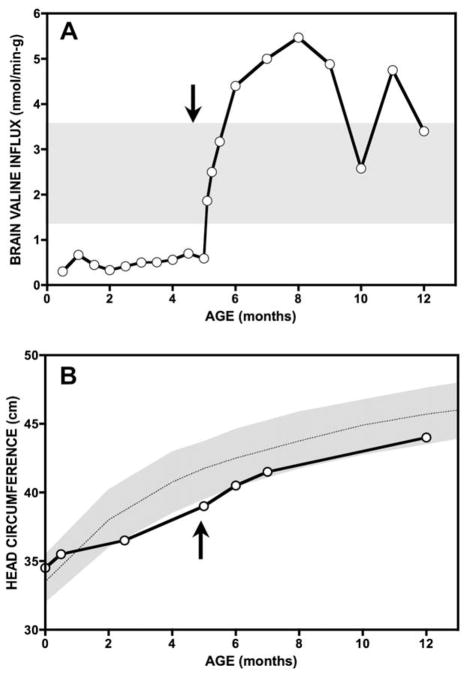
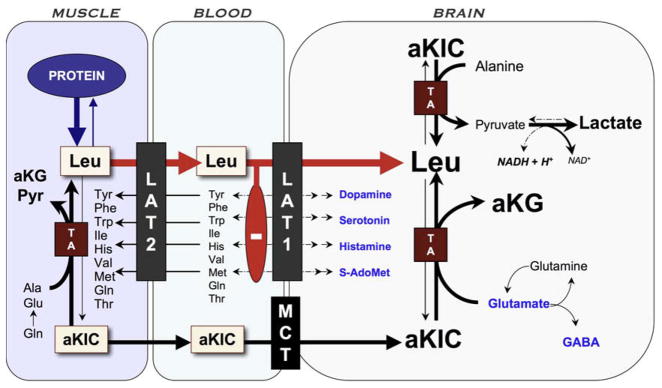
Branched-chain ketoacid dehydrogenase deficiency results in complex and volatile metabolic derangements that threaten brain development. Treatment for classical maple syrup urine disease (MSUD) should address this underlying physiology while also protecting children from nutrient deficiencies. Based on a 20-year experience managing 79 patients, we designed a study formula to (1) optimize transport of seven amino acids (Tyr, Trp, His, Met, Thr, Gln, Phe) that compete with branched-chain amino acids (BCAAs) for entry into the brain via a common transporter (LAT1), (2) compensate for episodic depletions of glutamine, glutamate, and alanine caused by reverse transamination, and (3) correct deficiencies of omega-3 essential fatty acids, zinc, and selenium widespread among MSUD patients. The formula was enriched with LAT1 amino acid substrates, glutamine, alanine, zinc, selenium, and alpha-linolenic acid (18:3n-3). Fifteen Old Order Mennonite children were started on study formula between birth and 34 months of age and seen at least monthly in the office. Amino acid levels were checked once weekly and more often during illnesses. All children grew and developed normally over a period of 14-33 months. Energy demand, leucine tolerance, and protein accretion were tightly linked during periods of normal growth. Rapid shifts to net protein degradation occurred during illnesses. At baseline, most LAT1 substrates varied inversely with plasma leucine, and their calculated rates of brain uptake were 20-68% below normal. Treatment with study formula increased plasma concentrations of LAT1 substrates and normalized their calculated uptakes into the nervous system. Red cell membrane omega-3 polyunsaturated fatty acids and serum zinc and selenium levels increased on study formula. However, selenium and docosahexaenoic acid (22:6n-3) levels remained below normal. During the study period, hospitalizations decreased from 0.35 to 0.14 per patient per year. There were 28 hospitalizations managed with MSUD hyperalimentation solution; 86% were precipitated by common infections, especially vomiting and gastroenteritis. The large majority of catabolic illnesses were managed successfully at home using 'sick-day' formula and frequent amino acid monitoring. We conclude that the study formula is safe and effective for the treatment of classical MSUD. In principle, dietary enrichment protects the brain against deficiency of amino acids used for protein accretion, neurotransmitter synthesis, and methyl group transfer. Although the pathophysiology of MSUD can be addressed through rational formula design, this does not replace the need for vigilant clinical monitoring, frequent measurement of the complete amino acid profile, and ongoing dietary adjustments that match nutritional intake to the metabolic demands of growth and illness.
Copyright 2009 Elsevier Inc. All rights reserved.
Figures
References
-
- Strauss KA, Morton DH. Branched-chain ketoacyl dehydrogenase deficiency: maple syrup disease. Curr Treat Options Neurol. 2003;5:329–341. - PubMed
-
- Morton DH, Morton CS, Strauss KA, Robinson DL, Puffenberger EG, Hendrickson C, Kelley RI. Pediatric medicine and the genetic disorders of the Amish and Mennonite people of Pennsylvania. Am J Med Genet C Semin Med Genet. 2003;121:5–17. - PubMed
-
- Strauss KA, Puffenberger EG, Morton DH. GeneReviews. University of Washington; Seattle: 2006. Maple Syrup Urine Disease. - PubMed
-
- Kamei A, Takashima S, Chan F, Becker LE. Abnormal dendritic development in maple syrup urine disease. Pediatr Neurol. 1992;8:145–147. - PubMed
-
- Smith QR, Stoll JS. Blood–brain barrier amino acid transport. In: Pardridge WM, editor. Introduction to the Blood–Brain Barrier. Cambridge University Press; Cambridge: 1998. pp. 188–197.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
