

Modulation of mitochondrial function and morphology by interaction of Omi/HtrA2 with the mitochondrial fusion factor OPA1
- PMID: 20064504
- PMCID: PMC3063334
- DOI: 10.1016/j.yexcr.2010.01.005
Modulation of mitochondrial function and morphology by interaction of Omi/HtrA2 with the mitochondrial fusion factor OPA1
Abstract
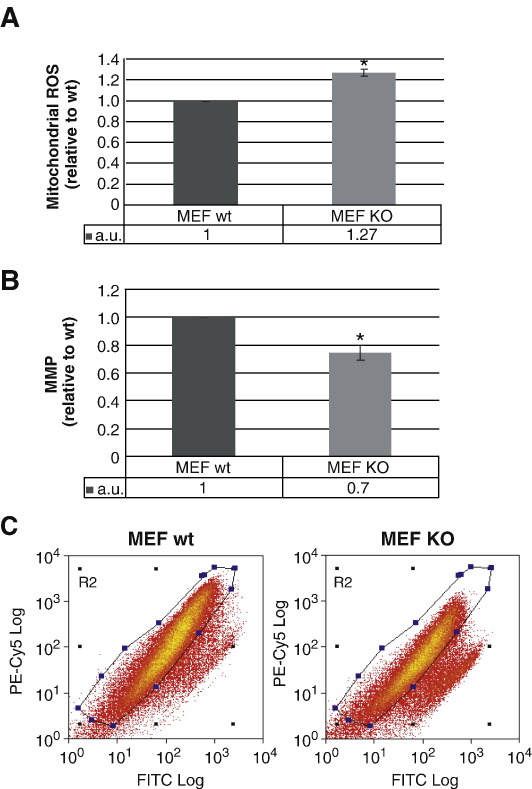
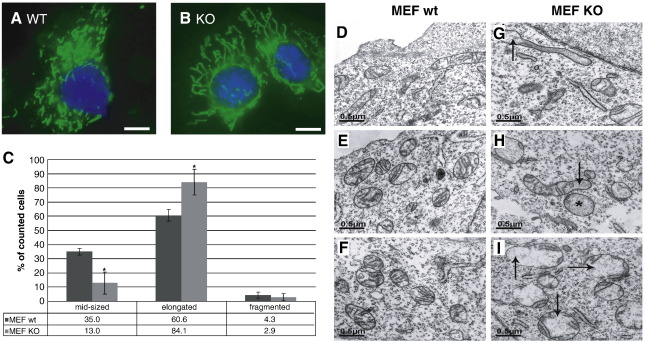
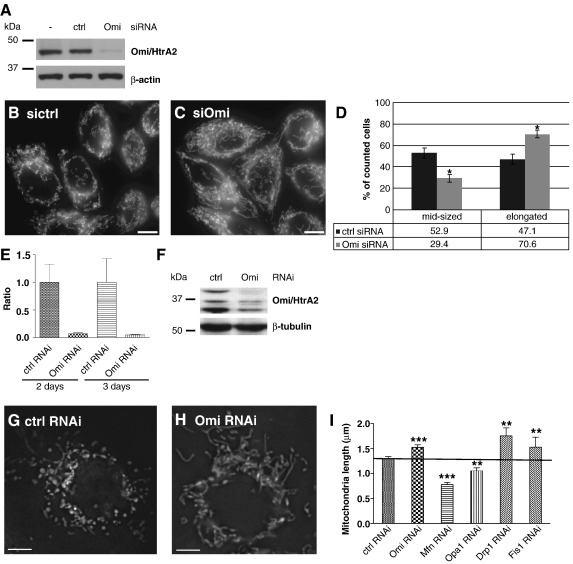
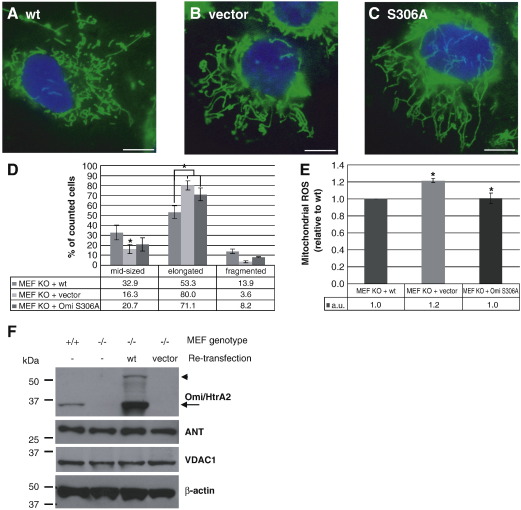
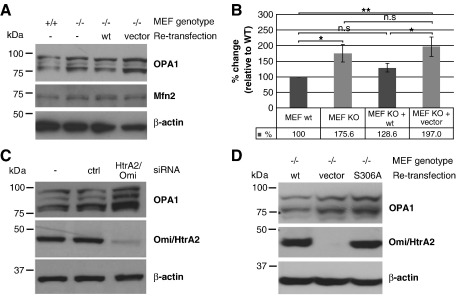
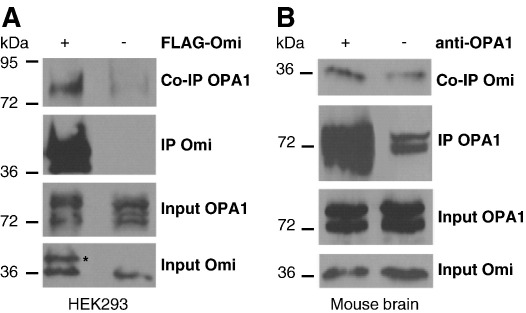
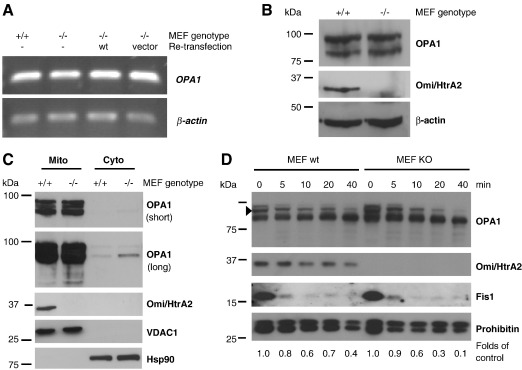
Loss of Omi/HtrA2 function leads to nerve cell loss in mouse models and has been linked to neurodegeneration in Parkinson's and Huntington's disease. Omi/HtrA2 is a serine protease released as a pro-apoptotic factor from the mitochondrial intermembrane space into the cytosol. Under physiological conditions, Omi/HtrA2 is thought to be involved in protection against cellular stress, but the cytological and molecular mechanisms are not clear. Omi/HtrA2 deficiency caused an accumulation of reactive oxygen species and reduced mitochondrial membrane potential. In Omi/HtrA2 knockout mouse embryonic fibroblasts, as well as in Omi/HtrA2 silenced human HeLa cells and Drosophila S2R+ cells, we found elongated mitochondria by live cell imaging. Electron microscopy confirmed the mitochondrial morphology alterations and showed abnormal cristae structure. Examining the levels of proteins involved in mitochondrial fusion, we found a selective up-regulation of more soluble OPA1 protein. Complementation of knockout cells with wild-type Omi/HtrA2 but not with the protease mutant [S306A]Omi/HtrA2 reversed the mitochondrial elongation phenotype and OPA1 alterations. Finally, co-immunoprecipitation showed direct interaction of Omi/HtrA2 with endogenous OPA1. Thus, we show for the first time a direct effect of loss of Omi/HtrA2 on mitochondrial morphology and demonstrate a novel role of this mitochondrial serine protease in the modulation of OPA1. Our results underscore a critical role of impaired mitochondrial dynamics in neurodegenerative disorders.
Copyright 2010 Elsevier Inc. All rights reserved.
Figures
References
-
- Vande Walle L., Lamkanfi M., Vandenabeele P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ. 2008;15:453–460. - PubMed
-
- Hegde R., Srinivasula S.M., Zhang Z., Wassell R., Mukattash R., Cilenti L., DuBois G., Lazebnik Y., Zervos A.S., Fernandes-Alnemri T., Alnemri E.S. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol Chem. 2002;277:432–438. - PubMed
-
- Martins L.M., Iaccarino I., Tenev T., Gschmeissner S., Totty N.F., Lemoine N.R., Savopoulos J., Gray C.W., Creasy C.L., Dingwall C., Downward J. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J. Biol Chem. 2002;277:439–444. - PubMed
-
- Verhagen A.M., Silke J., Ekert P.G., Pakusch M., Kaufmann H., Connolly L.M., Day C.L., Tikoo A., Burke R., Wrobel C., Moritz R.L., Simpson R.J., Vaux D.L. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J. Biol Chem. 2002;277:445–454. - PubMed
-
- Inagaki R., Tagawa K., Qi M.L., Enokido Y., Ito H., Tamura T., Shimizu S., Oyanagi K., Arai N., Kanazawa I., Wanker E.E., Okazawa H. Omi/HtrA2 is relevant to the selective vulnerability of striatal neurons in Huntington's disease. Eur. J. Neurosci. 2008;28:30–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
