

The "LEARn" (Latent Early-life Associated Regulation) model integrates environmental risk factors and the developmental basis of Alzheimer's disease, and proposes remedial steps
- PMID: 20064601
- PMCID: PMC2881328
- DOI: 10.1016/j.exger.2010.01.001
The "LEARn" (Latent Early-life Associated Regulation) model integrates environmental risk factors and the developmental basis of Alzheimer's disease, and proposes remedial steps
Abstract
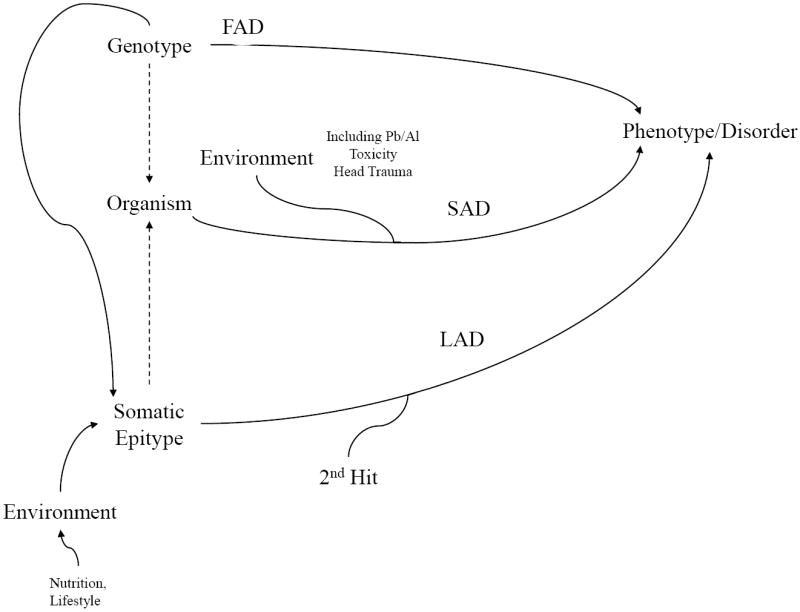
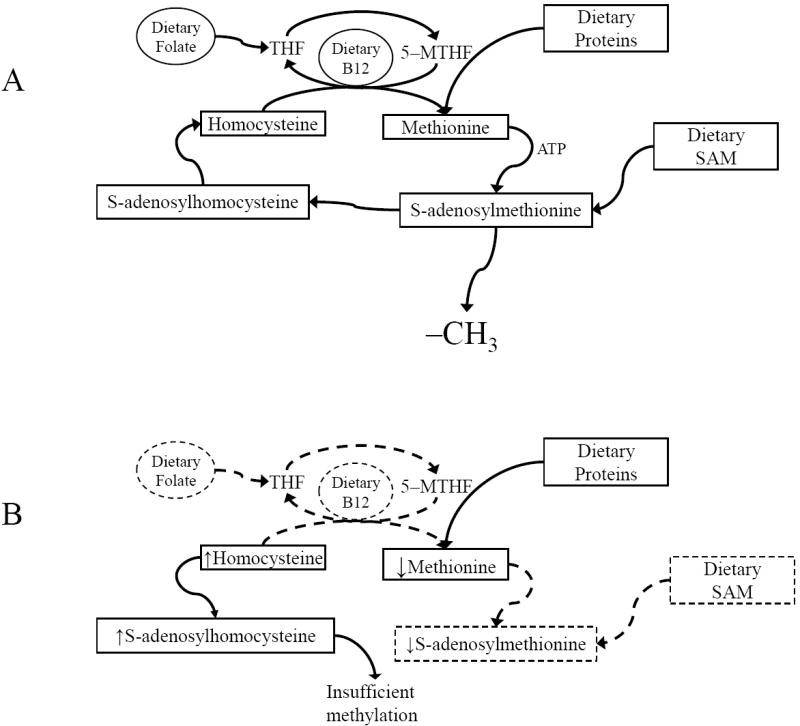
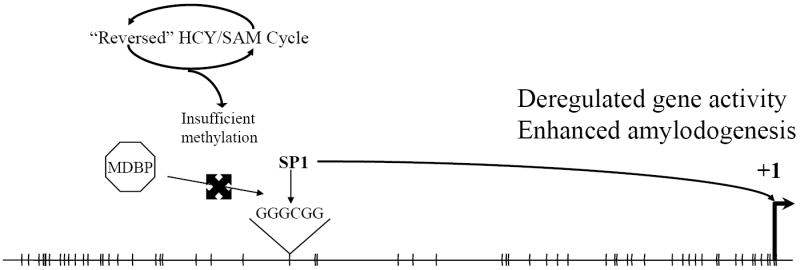
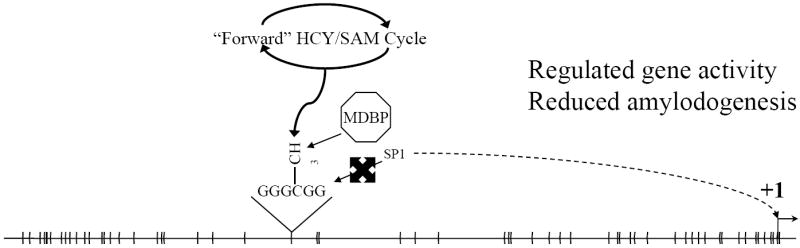
The neurodegenerative disorder Alzheimer's disease (AD) is the 6th leading cause of death in the USA. In addition to neurological and psychiatric symptoms, AD is characterized by deficiencies in S-adenylmethionine (SAM), vitamin B12, and folate. Deficiency in these nutrients has been shown to result in gene promoter methylation with upregulation of AD-associated genes. While some cases of AD are due to specific mutations in genes such as presenilin 1 (PSEN) and the amyloid-beta peptide precursor protein (APP), these familial AD (FAD) cases account for a minority of cases. The majority of genetic contribution consists of risk factors with incomplete penetrance. Several environmental risk factors, such as cholesterol and diet, head trauma, and reduced levels of exercise, have also been determined for AD. Nevertheless, the majority of risk for AD appears to be established early in life. We propose to explain this via the LEARn (Latent Early-life Associated Regulation) model. LEARn-AD (LAD) would be a "two-hit" disorder, wherein the first hit would occur due to environmental stress within the regulatory sequences of AD-associated genes, maintained by epigenetic changes such as in DNA methylation. This hit would most likely come in early childhood. The second hit could consist of further stress, such as head trauma, poor mid-life diet, or even general changes in expression of genes that occur later in life independent of any pathogenesis. Given that the primary risk for LAD would be maintained by DNA (hypo)methylation, we propose that long-term nutritional remediation based on the LEARn model, or LEARn-based nutritional gain (LEARnING), beginning early in life, would significantly reduce risk for AD late in life.
2010 Elsevier Inc. All rights reserved.
Figures
References
-
- Aisen PS, Egelko S, Andrews H, Diaz-Arrastia R, Weiner M, DeCarli C, Jagust W, Miller JW, Green R, Bell K, Sano M. A pilot study of vitamins to lower plasma homocysteine levels in Alzheimer disease. Am J Geriatr Psychiatry. 2003;11:246–9. - PubMed
-
- Alzheimer’s Association. 2009 Alzheimer’s disease facts and figures. Alzheimers Dement. 2009;5:234–70. - PubMed
-
- Ashford JW, Mortimer JA. Non-familial Alzheimer’s disease is mainly due to genetic factors. J Alzheimers Dis. 2002;4:169–77. - PubMed
-
- Bales KR, Du Y, Holtzman D, Cordell B, Paul SM. Neuroinflammation and Alzheimer’s disease: critical roles for cytokine/Abeta-induced glial activation, NF-kappaB, and apolipoprotein E. Neurobiology of Aging. 2000;21:427–432. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
