

Exogenous Nef is an inhibitor of Mycobacterium tuberculosis-induced tumor necrosis factor-alpha production and macrophage apoptosis
- PMID: 20068037
- PMCID: PMC2857058
- DOI: 10.1074/jbc.M109.073320
Exogenous Nef is an inhibitor of Mycobacterium tuberculosis-induced tumor necrosis factor-alpha production and macrophage apoptosis
Erratum in
-
Exogenous Nef is an inhibitor of Mycobacterium tuberculosis -induced tumor necrosis factor-α production and macrophage apoptosis.J Biol Chem. 2016 Jan 8;291(2):665-6. doi: 10.1074/jbc.A109.073320. J Biol Chem. 2016. PMID: 26747889 Free PMC article. No abstract available.
Abstract
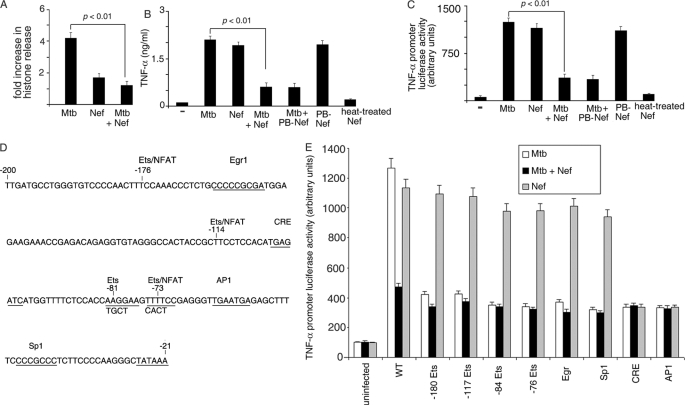
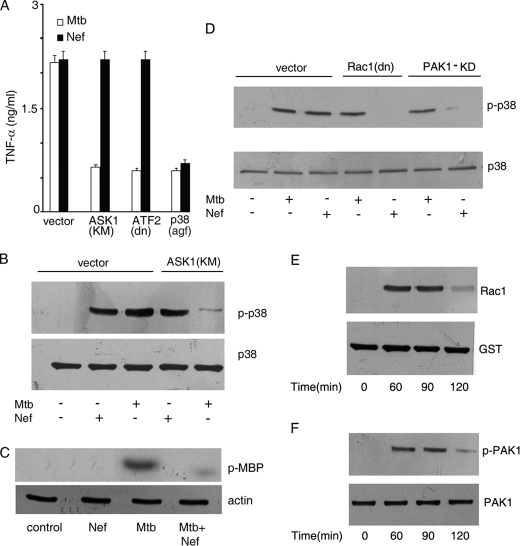
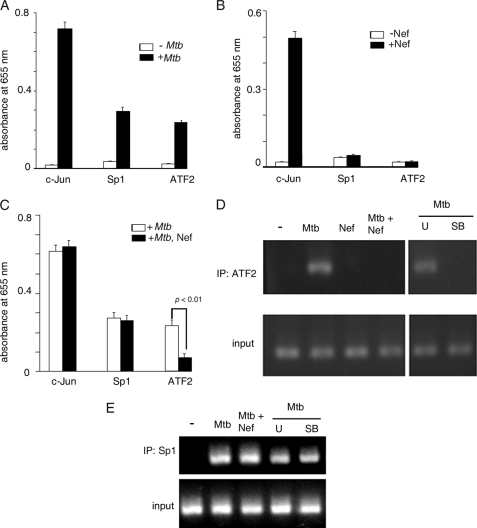
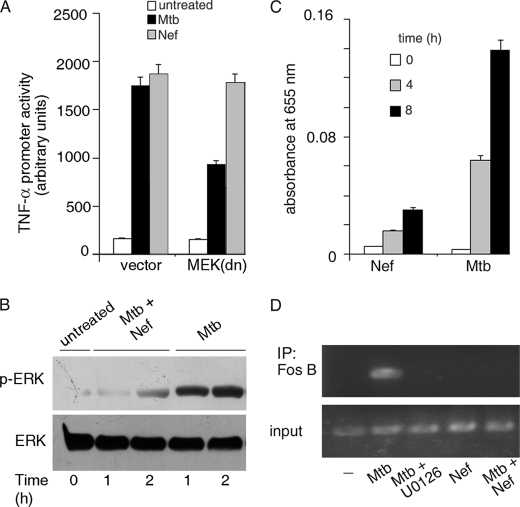
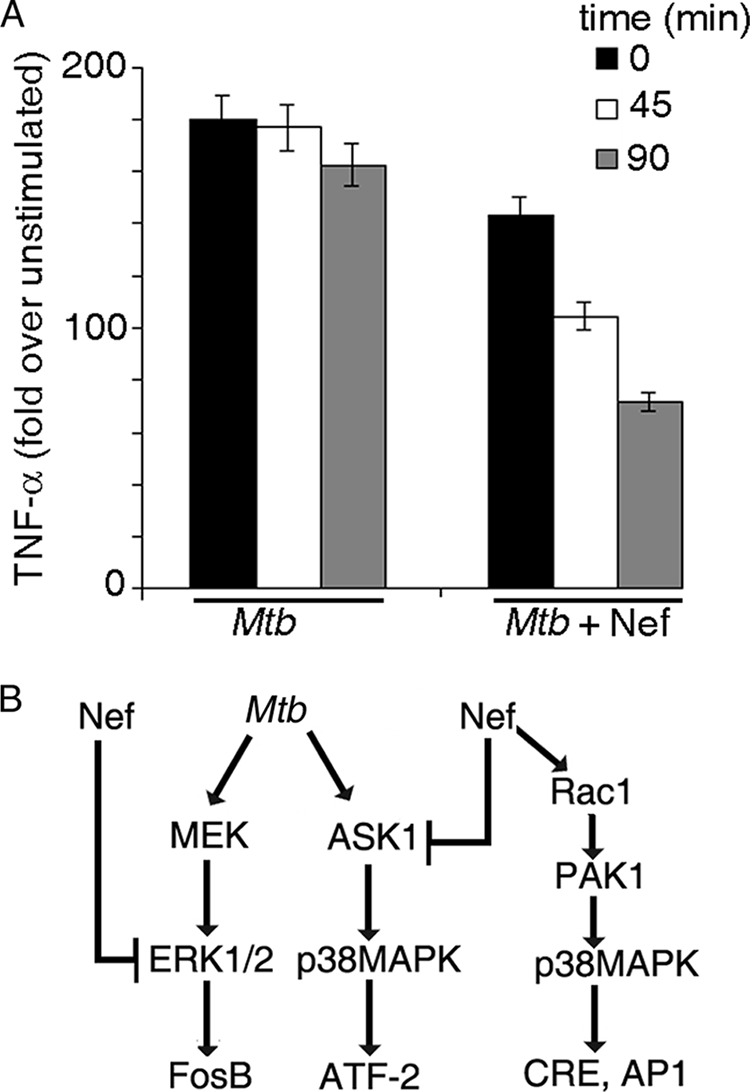
Human immunodeficiency virus-1 (HIV-1) impairs tumor necrosis factor-alpha (TNF-alpha)-mediated macrophage apoptosis induced by Mycobacterium tuberculosis (Mtb). HIV Nef protein plays an important role in the pathogenesis of AIDS. We have tested the hypothesis that exogenous Nef is a factor that inhibits TNF-alpha production/apoptosis in macrophages infected with Mtb. We demonstrate that Mtb and Nef individually trigger TNF-alpha production in macrophages. However, TNF-alpha production is dampened when the two are present simultaneously, probably through cross-regulation of the individual signaling pathways leading to activation of the TNF-alpha promoter. Mtb-induced TNF-alpha production is abrogated upon mutation of the Ets, Egr, Sp1, CRE, or AP1 binding sites on the TNF-alpha promoter, whereas Nef-mediated promoter activation depends only on the CRE and AP1 binding sites, pointing to differences in the mechanisms of activation of the promoter. Mtb-dependent promoter activation depends on the mitogen-activated kinase (MAPK) kinase kinase ASK1 and on MEK/ERK signaling. Nef inhibits ASK1/p38 MAPK-dependent Mtb-induced TNF-alpha production probably by inhibiting binding of ATF2 to the TNF-alpha promoter. It also inhibits MEK/ERK-dependent Mtb-induced binding of FosB to the promoter. Nef-driven TNF-alpha production occurs in an ASK1-independent, Rac1/PAK1/p38 MAPK-dependent, and MEK/ERK-independent manner. The signaling pathways used by Mtb and Nef to trigger TNF-alpha production are therefore distinctly different. In addition to attenuating Mtb-dependent TNF-alpha promoter activation, Nef also reduces Mtb-dependent TNF-alpha mRNA stability probably through its ability to inhibit ASK1/p38 MAPK signaling. These results provide new insight into how HIV Nef probably exacerbates tuberculosis infection by virtue of its ability to dampen Mtb-induced TNF-alpha production.
Figures
Similar articles
-
Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-alpha.J Biol Chem. 2007 Jan 12;282(2):1039-50. doi: 10.1074/jbc.M604379200. Epub 2006 Nov 9. J Biol Chem. 2007. PMID: 17095513
-
Mycobacterium tuberculosis EsxL induces TNF-α secretion through activation of TLR2 dependent MAPK and NF-κB pathways.Mol Immunol. 2021 Feb;130:133-141. doi: 10.1016/j.molimm.2020.11.020. Epub 2021 Jan 6. Mol Immunol. 2021. PMID: 33419561
-
Pre-exposure of Mycobacterium tuberculosis-infected macrophages to crystalline silica impairs control of bacterial growth by deregulating the balance between apoptosis and necrosis.PLoS One. 2013 Nov 22;8(11):e80971. doi: 10.1371/journal.pone.0080971. eCollection 2013. PLoS One. 2013. PMID: 24278357 Free PMC article.
-
Insights into battles between Mycobacterium tuberculosis and macrophages.Protein Cell. 2014 Oct;5(10):728-36. doi: 10.1007/s13238-014-0077-5. Epub 2014 Jun 18. Protein Cell. 2014. PMID: 24938416 Free PMC article. Review.
-
Macrophage signaling in HIV-1 infection.Retrovirology. 2010 Apr 9;7:34. doi: 10.1186/1742-4690-7-34. Retrovirology. 2010. PMID: 20380698 Free PMC article. Review.
Cited by
-
Uncovering the Bronchoalveolar Single-Cell Landscape of Patients With Pulmonary Tuberculosis With Human Immunodeficiency Virus Type 1 Coinfection.J Infect Dis. 2024 Sep 23;230(3):e524-e535. doi: 10.1093/infdis/jiae042. J Infect Dis. 2024. PMID: 38412342 Free PMC article.
-
Protein phosphatase, Mg2+/Mn2+-dependent 1A controls the innate antiviral and antibacterial response of macrophages during HIV-1 and Mycobacterium tuberculosis infection.Oncotarget. 2016 Mar 29;7(13):15394-409. doi: 10.18632/oncotarget.8190. Oncotarget. 2016. PMID: 27004401 Free PMC article.
-
Multifaceted Impact of Host C-C Chemokine CCL2 in the Immuno-Pathogenesis of HIV-1/M. tuberculosis Co-Infection.Front Immunol. 2013 Oct 4;4:312. doi: 10.3389/fimmu.2013.00312. Front Immunol. 2013. PMID: 24109479 Free PMC article. Review.
-
HIV Nef Expression Down-modulated GFAP Expression and Altered Glutamate Uptake and Release and Proliferation in Astrocytes.Aging Dis. 2023 Feb 1;14(1):152-169. doi: 10.14336/AD.2022.0712. eCollection 2023 Feb 1. Aging Dis. 2023. PMID: 36818564 Free PMC article.
-
Signaling through the P38 and ERK pathways: a common link between HIV replication and the immune response.Immunol Res. 2010 Dec;48(1-3):99-109. doi: 10.1007/s12026-010-8170-1. Immunol Res. 2010. PMID: 20725863 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
