

Cardiomyocyte PDGFR-beta signaling is an essential component of the mouse cardiac response to load-induced stress
- PMID: 20071776
- PMCID: PMC2810076
- DOI: 10.1172/JCI39434
Cardiomyocyte PDGFR-beta signaling is an essential component of the mouse cardiac response to load-induced stress
Abstract
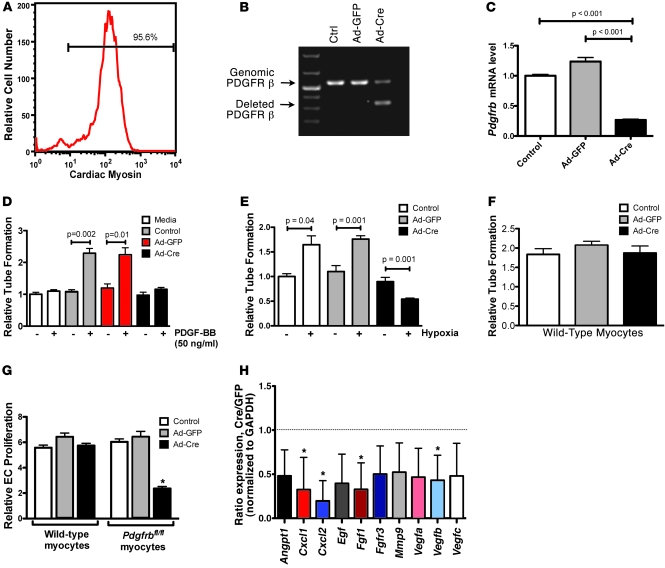
PDGFR is an important target for novel anticancer therapeutics because it is overexpressed in a wide variety of malignancies. Recently, however, several anticancer drugs that inhibit PDGFR signaling have been associated with clinical heart failure. Understanding this effect of PDGFR inhibitors has been difficult because the role of PDGFR signaling in the heart remains largely unexplored. As described herein, we have found that PDGFR-beta expression and activation increase dramatically in the hearts of mice exposed to load-induced cardiac stress. In mice in which Pdgfrb was knocked out in the heart in development or in adulthood, exposure to load-induced stress resulted in cardiac dysfunction and heart failure. Mechanistically, we showed that cardiomyocyte PDGFR-beta signaling plays a vital role in stress-induced cardiac angiogenesis. Specifically, we demonstrated that cardiomyocyte PDGFR-beta was an essential upstream regulator of the stress-induced paracrine angiogenic capacity (the angiogenic potential) of cardiomyocytes. These results demonstrate that cardiomyocyte PDGFR-beta is a regulator of the compensatory cardiac response to pressure overload-induced stress. Furthermore, our findings may provide insights into the mechanism of cardiotoxicity due to anticancer PDGFR inhibitors.
Figures






References
-
- Ostman A, Heldin CH. Involvement of platelet-derived growth factor in disease: development of specific antagonists. Adv Cancer Res. 2001;80:1–38. - PubMed
-
- Uehara H, et al. Effects of blocking platelet-derived growth factor-receptor signaling in a mouse model of experimental prostate cancer bone metastases. . J Natl Cancer Inst. 2003;95(6):458–470. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
