

A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping
- PMID: 20072625
- PMCID: PMC2800183
- DOI: 10.1371/journal.pone.0008647
A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping
Abstract
Background: Duchenne muscular dystrophy (DMD), which afflicts 1 in 3500 boys, is one of the most common genetic disorders of children. This fatal degenerative condition is caused by an absence or deficiency of dystrophin in striated muscle. Most affected patients have inherited or spontaneous deletions in the dystrophin gene that disrupt the reading frame resulting in unstable truncated products. For these patients, restoration of the reading frame via antisense oligonucleotide-mediated exon skipping is a promising therapeutic approach. The major DMD deletion "hot spot" is found between exons 45 and 53, and skipping exon 51 in particular is predicted to ameliorate the dystrophic phenotype in the greatest number of patients. Currently the mdx mouse is the most widely used animal model of DMD, although its mild phenotype limits its suitability in clinical trials. The Golden Retriever muscular dystrophy (GRMD) model has a severe phenotype, but due to its large size, is expensive to use. Both these models have mutations in regions of the dystrophin gene distant from the commonly mutated DMD "hot spot".
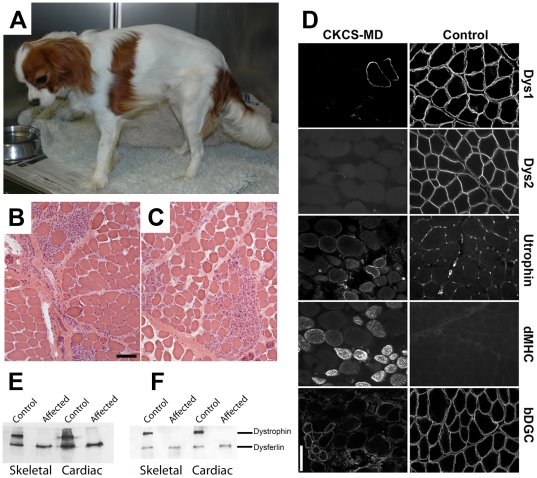
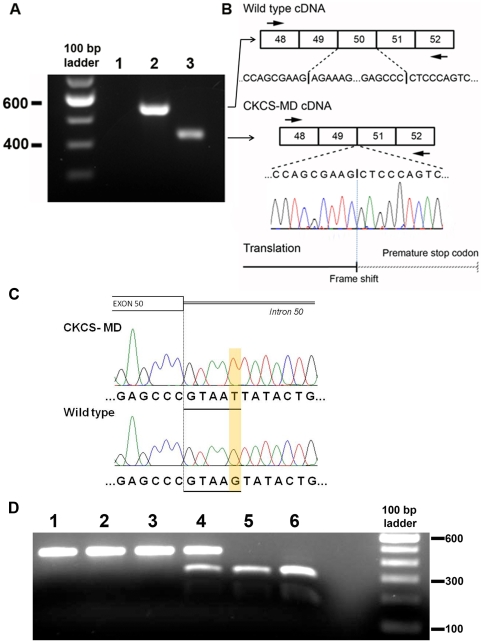
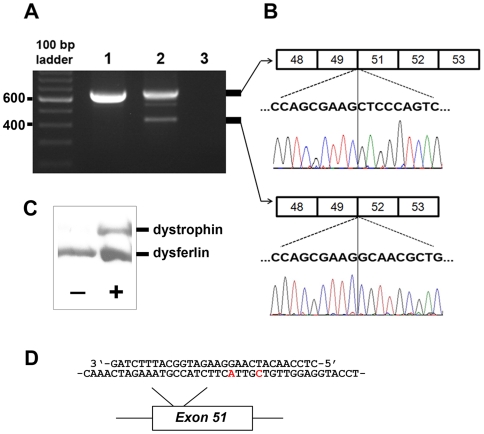
Methodology/principal findings: Here we describe the severe phenotype, histopathological findings, and molecular analysis of Cavalier King Charles Spaniels with dystrophin-deficient muscular dystrophy (CKCS-MD). The dogs harbour a missense mutation in the 5' donor splice site of exon 50 that results in deletion of exon 50 in mRNA transcripts and a predicted premature truncation of the translated protein. Antisense oligonucleotide-mediated skipping of exon 51 in cultured myoblasts from an affected dog restored the reading frame and protein expression.
Conclusions/significance: Given the small size of the breed, the amiable temperament and the nature of the mutation, we propose that CKCS-MD is a valuable new model for clinical trials of antisense oligonucleotide-induced exon skipping and other therapeutic approaches for DMD.
Conflict of interest statement
Figures
Similar articles
-
Biochemical characterization of patients with in-frame or out-of-frame DMD deletions pertinent to exon 44 or 45 skipping.JAMA Neurol. 2014 Jan;71(1):32-40. doi: 10.1001/jamaneurol.2013.4908. JAMA Neurol. 2014. PMID: 24217213 Clinical Trial.
-
In Vitro Multiexon Skipping by Antisense PMOs in Dystrophic Dog and Exon 7-Deleted DMD Patient.Methods Mol Biol. 2018;1828:151-163. doi: 10.1007/978-1-4939-8651-4_9. Methods Mol Biol. 2018. PMID: 30171540 Free PMC article.
-
In Vivo Evaluation of Multiple Exon Skipping with Peptide-PMOs in Cardiac and Skeletal Muscles in Dystrophic Dogs.Methods Mol Biol. 2018;1828:365-379. doi: 10.1007/978-1-4939-8651-4_23. Methods Mol Biol. 2018. PMID: 30171554
-
Skipping multiple exons of dystrophin transcripts using cocktail antisense oligonucleotides.Nucleic Acid Ther. 2014 Feb;24(1):57-68. doi: 10.1089/nat.2013.0451. Epub 2013 Dec 31. Nucleic Acid Ther. 2014. PMID: 24380394 Review.
-
Molecular correction of Duchenne muscular dystrophy by splice modulation and gene editing.RNA Biol. 2021 Jul;18(7):1048-1062. doi: 10.1080/15476286.2021.1874161. Epub 2021 Jan 20. RNA Biol. 2021. PMID: 33472516 Free PMC article. Review.
Cited by
-
X-Linked Duchenne-Type Muscular Dystrophy in Jack Russell Terrier Associated with a Partial Deletion of the Canine DMD Gene.Genes (Basel). 2020 Oct 8;11(10):1175. doi: 10.3390/genes11101175. Genes (Basel). 2020. PMID: 33049940 Free PMC article.
-
Identification of quantitative polymerase chain reaction reference genes suitable for normalising gene expression in the brain of normal and dystrophic mice and dogs.Wellcome Open Res. 2023 May 5;6:84. doi: 10.12688/wellcomeopenres.16707.2. eCollection 2021. Wellcome Open Res. 2023. PMID: 37942409 Free PMC article.
-
Evaluation of point mutations in dystrophin gene in Iranian Duchenne and Becker muscular dystrophy patients: introducing three novel variants.J Genet. 2016 Jun;95(2):325-9. doi: 10.1007/s12041-016-0641-2. J Genet. 2016. PMID: 27350676
-
Inhibition of CD26/DPP-IV enhances donor muscle cell engraftment and stimulates sustained donor cell proliferation.Skelet Muscle. 2012 Feb 16;2(1):4. doi: 10.1186/2044-5040-2-4. Skelet Muscle. 2012. PMID: 22340947 Free PMC article.
-
Mutation-Based Therapeutic Strategies for Duchenne Muscular Dystrophy: From Genetic Diagnosis to Therapy.J Pers Med. 2019 Mar 4;9(1):16. doi: 10.3390/jpm9010016. J Pers Med. 2019. PMID: 30836656 Free PMC article. Review.
References
-
- Bushby K, Bourke J, Bullock R, Eagle M, Gibson M, et al. The multidisciplinary management of Duchenne muscular dystrophy. Current Paediatrics. 2005;15:292–300.
-
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. - PubMed
-
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. - PubMed
-
- Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet. 1993;3:283–291. - PubMed
-
- Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006;7:762–773. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
