

Variable structure motifs for transcription factor binding sites
- PMID: 20074339
- PMCID: PMC2824720
- DOI: 10.1186/1471-2164-11-30
Variable structure motifs for transcription factor binding sites
Abstract
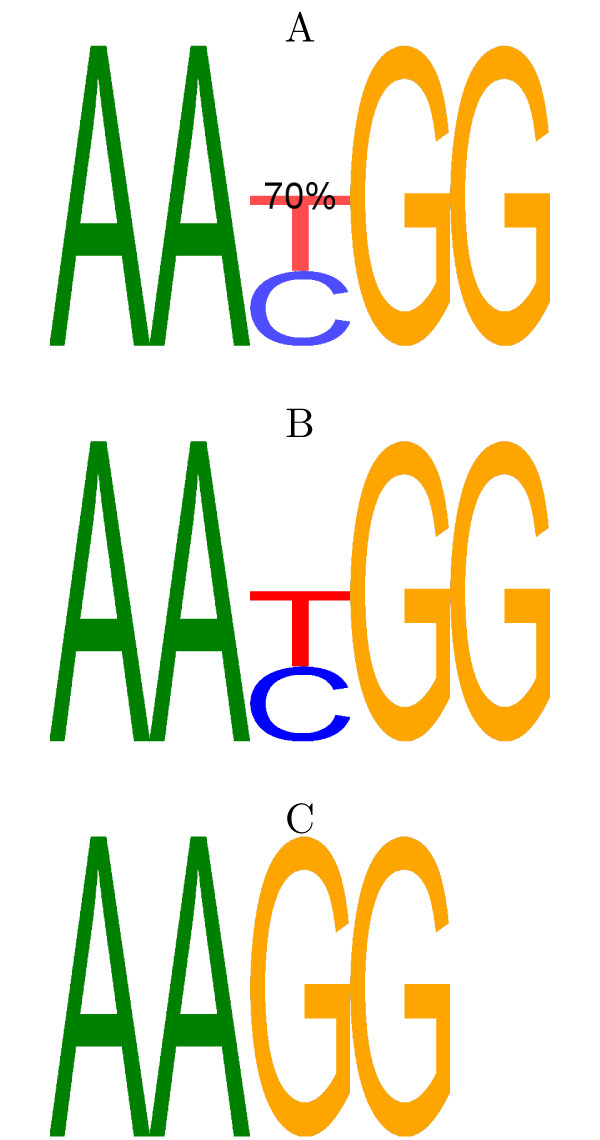
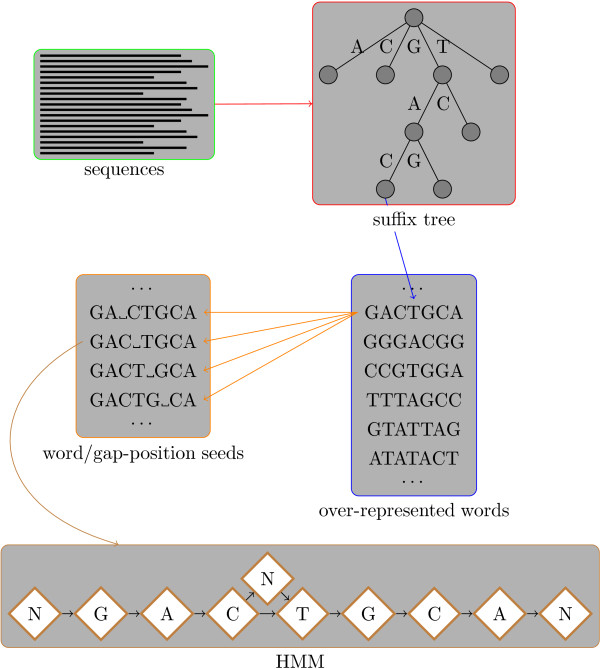
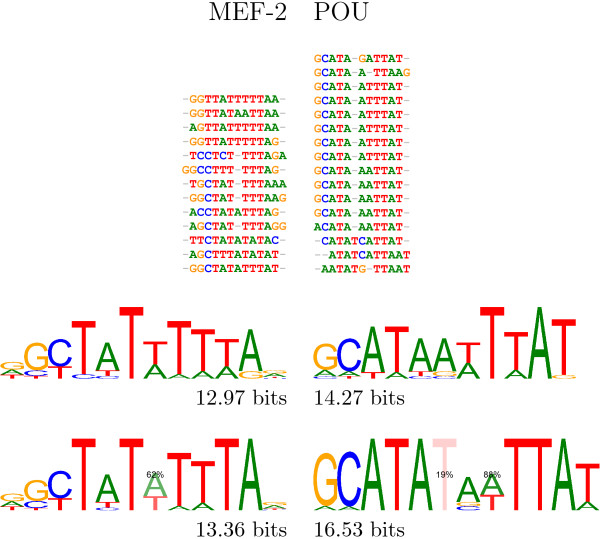
Background: Classically, models of DNA-transcription factor binding sites (TFBSs) have been based on relatively few known instances and have treated them as sites of fixed length using position weight matrices (PWMs). Various extensions to this model have been proposed, most of which take account of dependencies between the bases in the binding sites. However, some transcription factors are known to exhibit some flexibility and bind to DNA in more than one possible physical configuration. In some cases this variation is known to affect the function of binding sites. With the increasing volume of ChIP-seq data available it is now possible to investigate models that incorporate this flexibility. Previous work on variable length models has been constrained by: a focus on specific zinc finger proteins in yeast using restrictive models; a reliance on hand-crafted models for just one transcription factor at a time; and a lack of evaluation on realistically sized data sets.
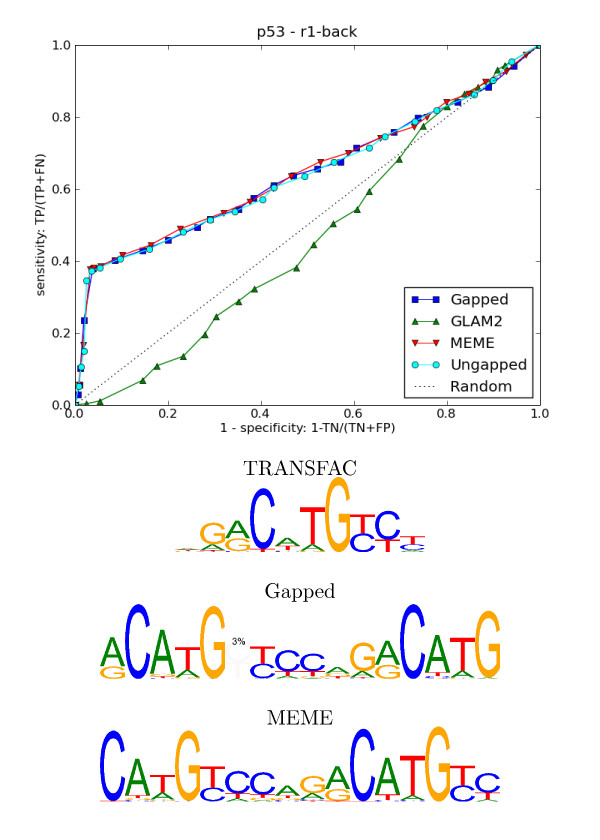
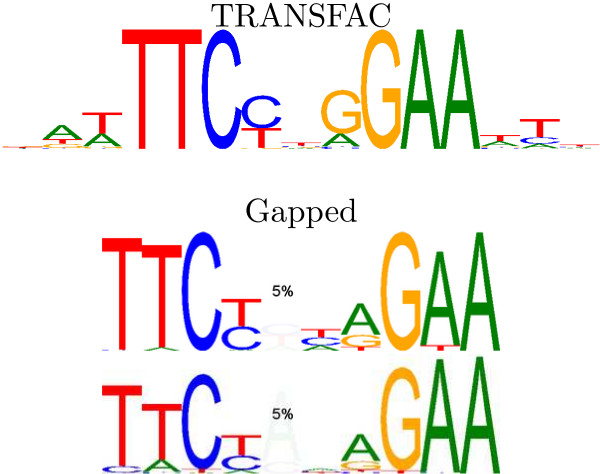
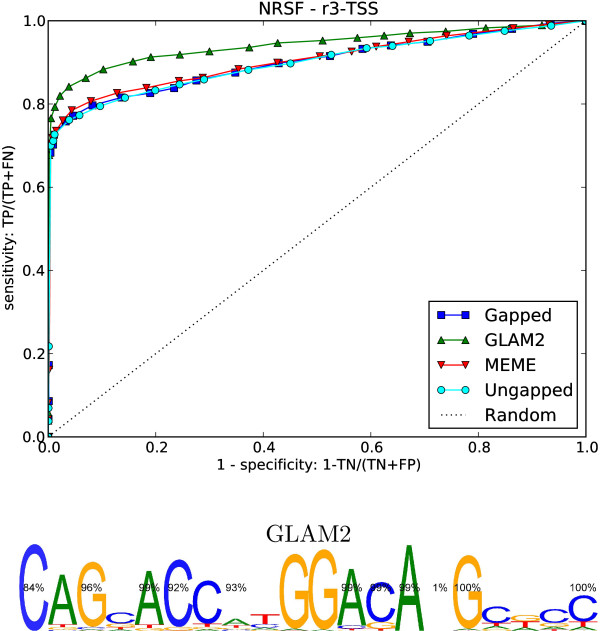
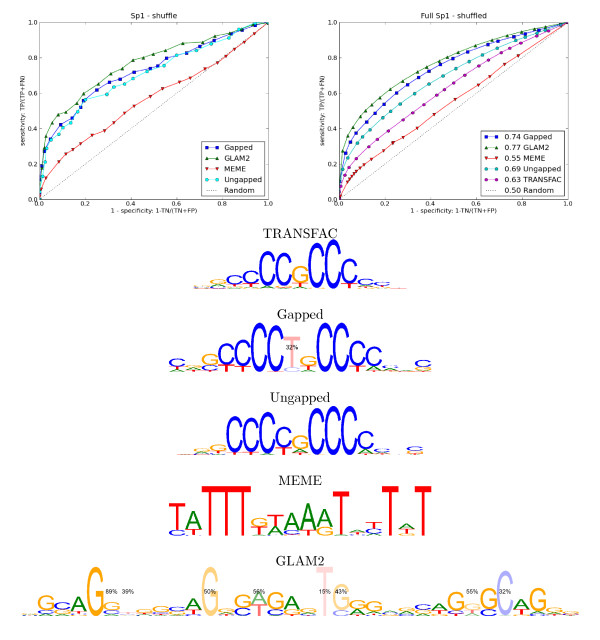
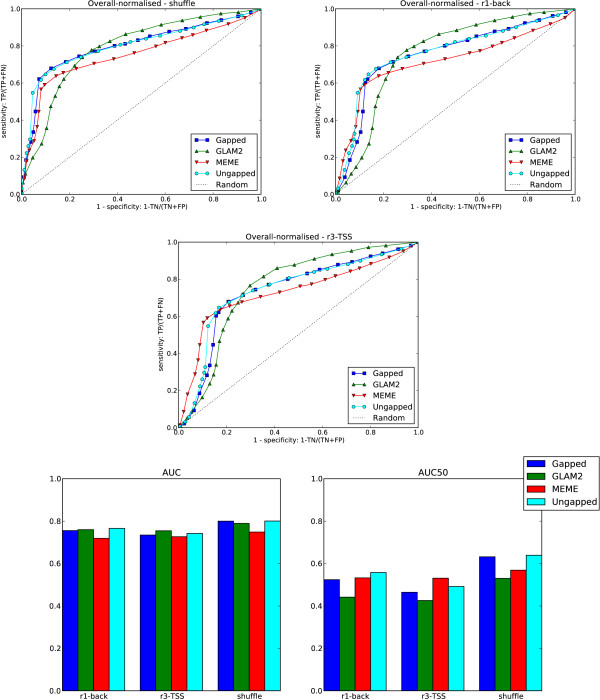
Results: We re-analysed binding sites from the TRANSFAC database and found motivating examples where our new variable length model provides a better fit. We analysed several ChIP-seq data sets with a novel motif search algorithm and compared the results to one of the best standard PWM finders and a recently developed alternative method for finding motifs of variable structure. All the methods performed comparably in held-out cross validation tests. Known motifs of variable structure were recovered for p53, Stat5a and Stat5b. In addition our method recovered a novel generalised version of an existing PWM for Sp1 that allows for variable length binding. This motif improved classification performance.
Conclusions: We have presented a new gapped PWM model for variable length DNA binding sites that is not too restrictive nor over-parameterised. Our comparison with existing tools shows that on average it does not have better predictive accuracy than existing methods. However, it does provide more interpretable models of motifs of variable structure that are suitable for follow-up structural studies. To our knowledge, we are the first to apply variable length motif models to eukaryotic ChIP-seq data sets and consequently the first to show their value in this domain. The results include a novel motif for the ubiquitous transcription factor Sp1.
Figures


References
-
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, Wong KY, Sung KW, Lee CW, Zhao XD, Chiu KP, Lipovich L, Kuznetsov VA, Robson P, Stanton LW, Wei CL, Ruan Y, Lim B, Ng HH. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38(4):431–40. doi: 10.1038/ng1760. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
