

Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis
- PMID: 20074522
- PMCID: PMC2885139
- DOI: 10.1016/j.cell.2009.12.024
Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis
Abstract
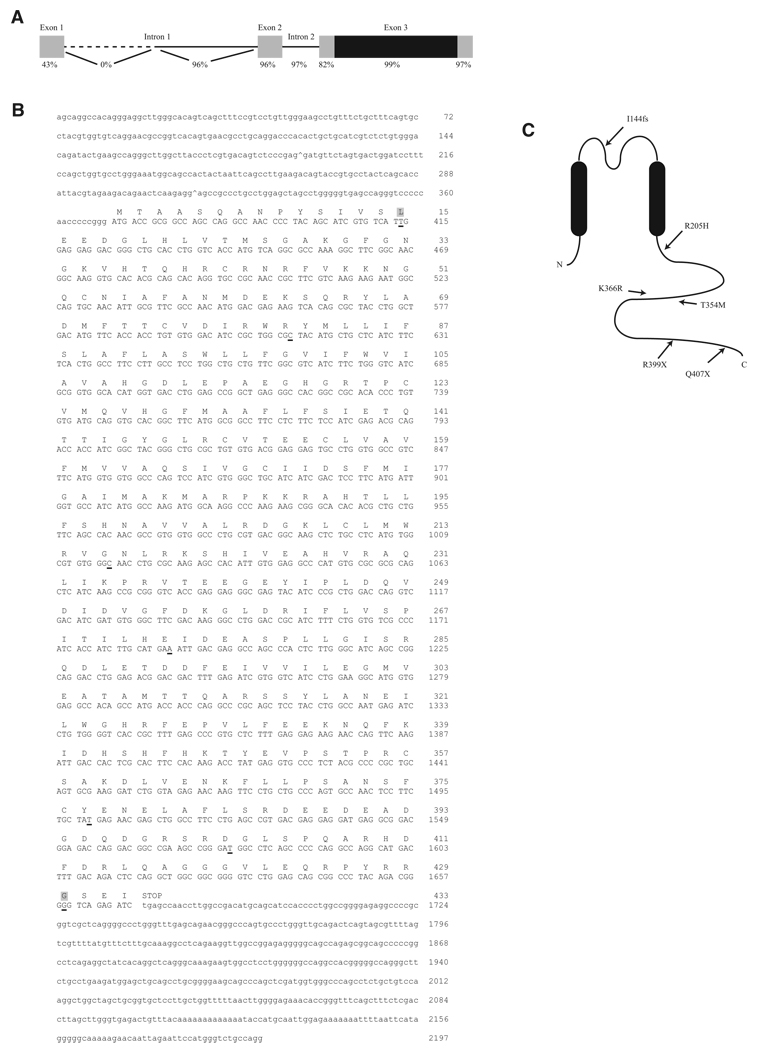
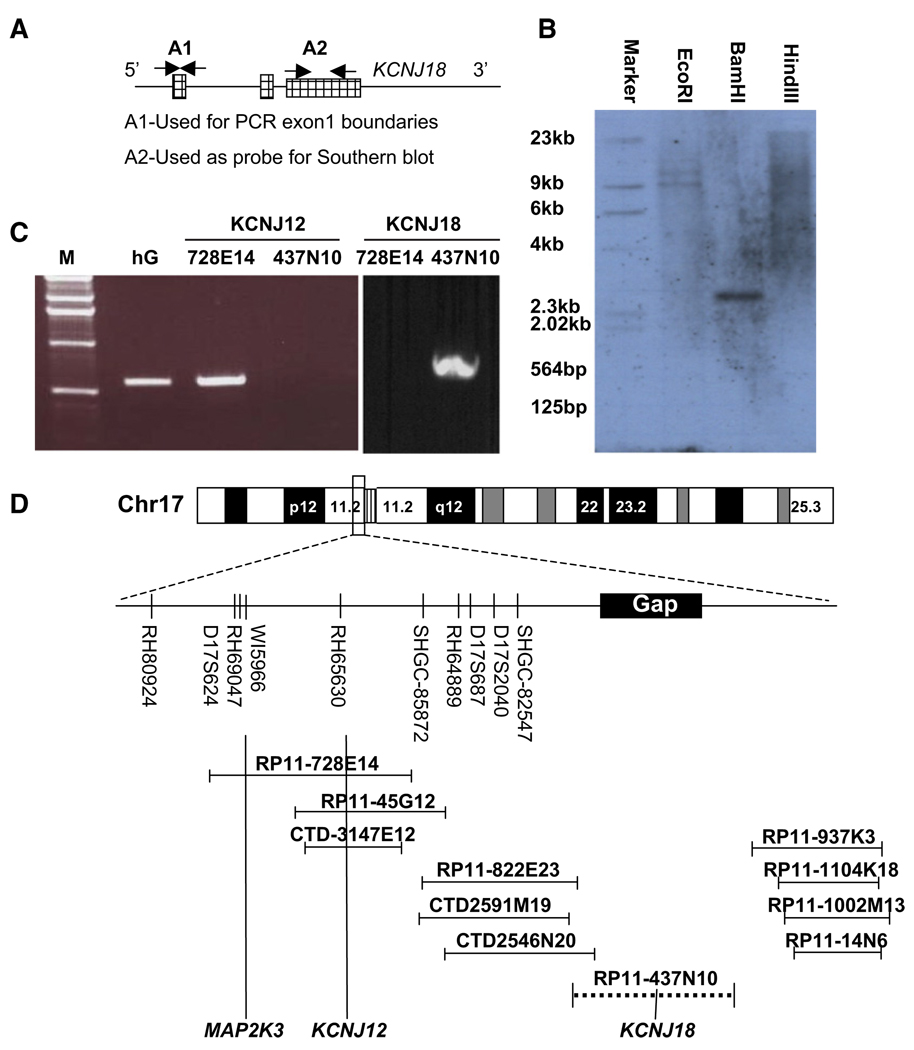
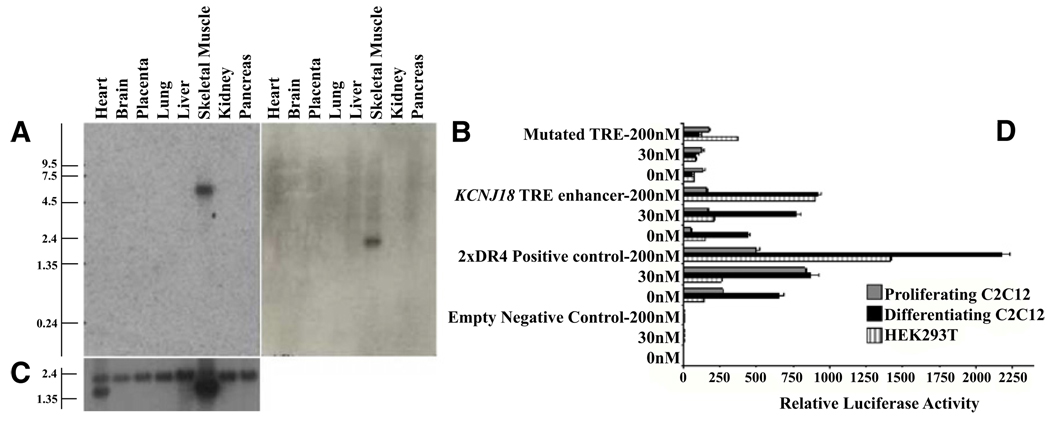
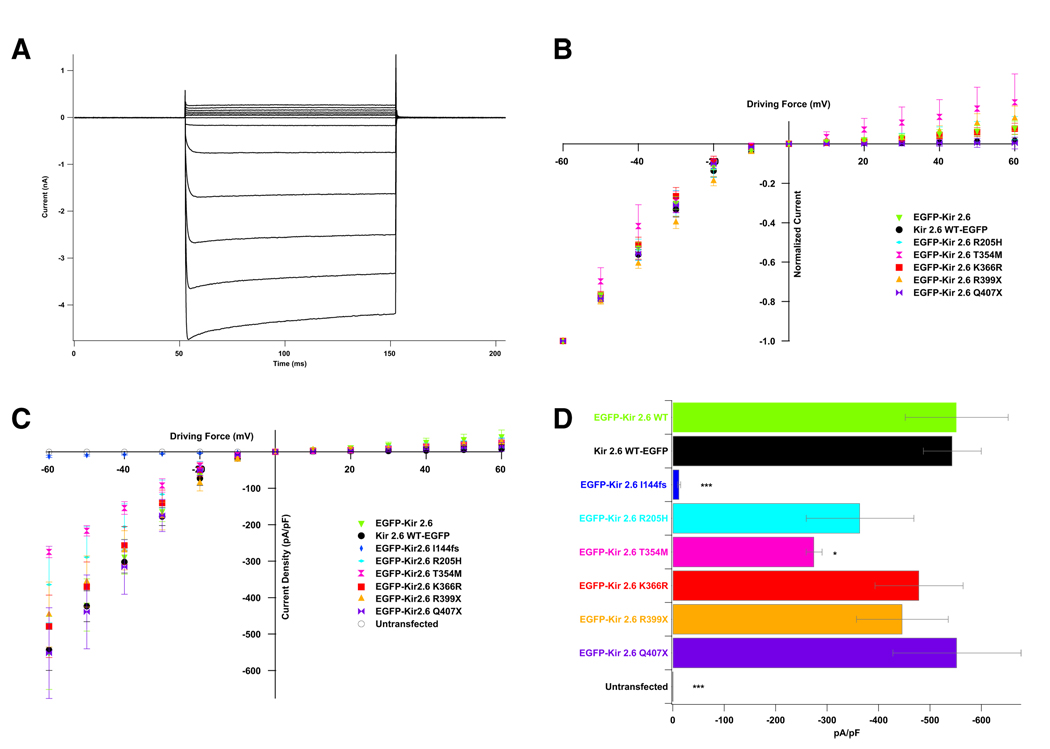
Thyrotoxic hypokalemic periodic paralysis (TPP) is characterized by acute attacks of weakness, hypokalemia, and thyrotoxicosis of various etiologies. These transient attacks resemble those of patients with familial hypokalemic periodic paralysis (hypoKPP) and resolve with treatment of the underlying hyperthyroidism. Because of the phenotypic similarity of these conditions, we hypothesized that TPP might also be a channelopathy. While sequencing candidate genes, we identified a previously unreported gene (not present in human sequence databases) that encodes an inwardly rectifying potassium (Kir) channel, Kir2.6. This channel, nearly identical to Kir2.2, is expressed in skeletal muscle and is transcriptionally regulated by thyroid hormone. Expression of Kir2.6 in mammalian cells revealed normal Kir currents in whole-cell and single-channel recordings. Kir2.6 mutations were present in up to 33% of the unrelated TPP patients in our collection. Some of these mutations clearly alter a variety of Kir2.6 properties, all altering muscle membrane excitability leading to paralysis.
Figures
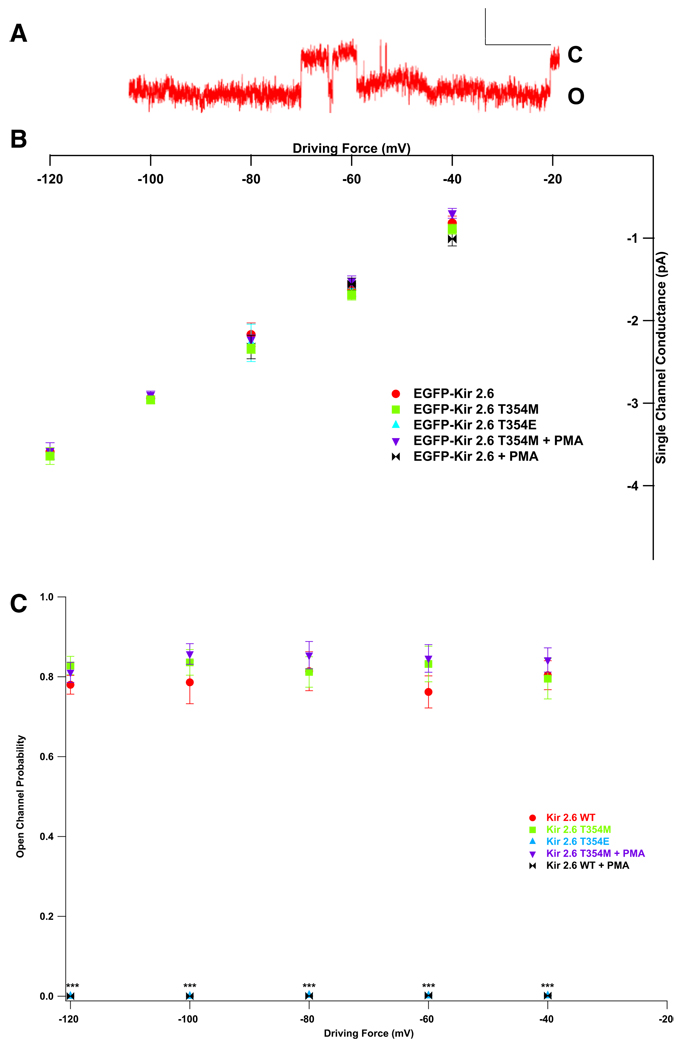
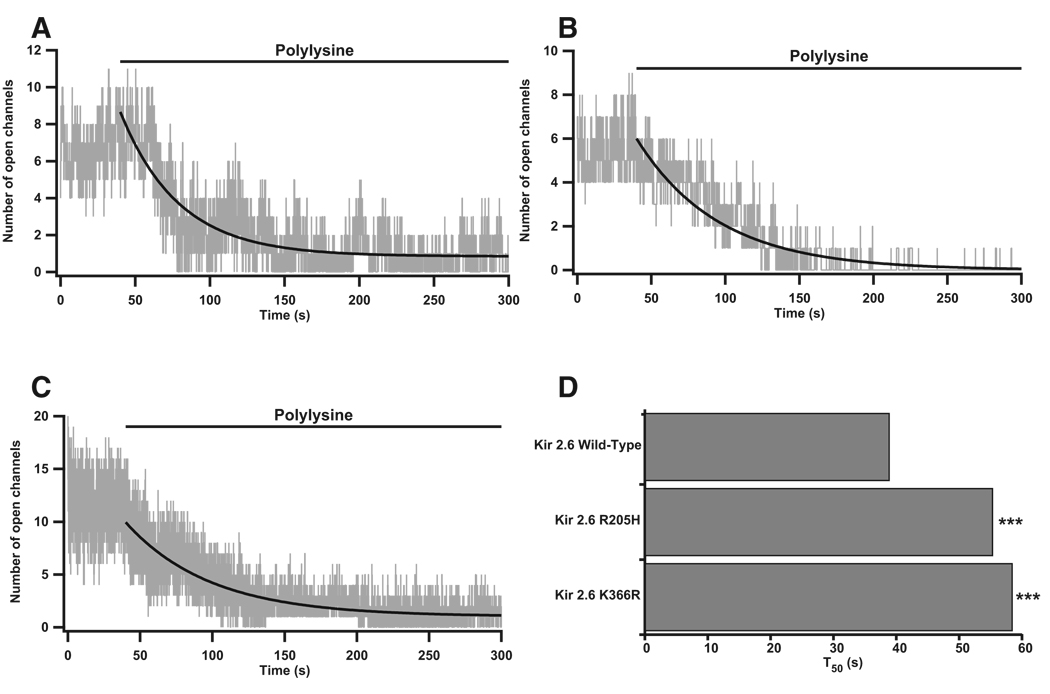
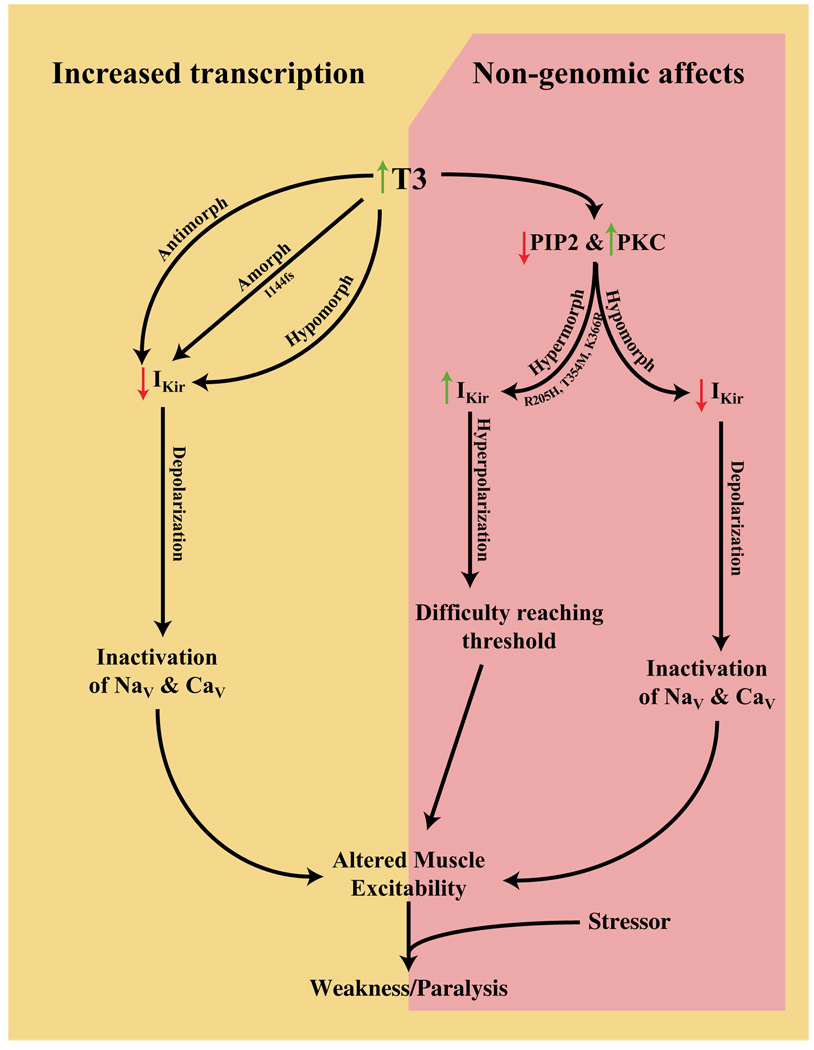
Comment in
-
'Kir'-ing thyrotoxic periodic paralysis.Clin Genet. 2010 Aug;78(2):136-8. doi: 10.1111/j.1399-0004.2010.01452_3.x. Clin Genet. 2010. PMID: 20662856 No abstract available.
References
-
- Abraham MR, Jahangir A, Alekseev AE, Terzic A. Channelopathies of inwardly rectifying potassium channels. FASEB J. 1999;13:1901–1910. - PubMed
-
- Bassett JH, Harvey CB, Williams GR. Mechanisms of thyroid hormone receptor-specific nuclear and extra nuclear actions. Mol. Cell. Endocrinol. 2003;213:1–11. - PubMed
-
- Bulman DE, Scoggan KA, van Oene MD, Nicolle MW, Hahn AF, Tollar LL, Ebers GC. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology. 1999;53:1932–1936. - PubMed
-
- Donaldson MR, Jensen JL, Tristani-Firouzi M, Tawil R, Bendahhou S, Suarez WA, Cobo AM, Poza JJ, Behr E, Wagstaff J, et al. PIP2 binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology. 2003;60:1811–1816. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
