

Cdc2-like kinase 2 is an insulin-regulated suppressor of hepatic gluconeogenesis
- PMID: 20074525
- PMCID: PMC2807620
- DOI: 10.1016/j.cmet.2009.11.006
Cdc2-like kinase 2 is an insulin-regulated suppressor of hepatic gluconeogenesis
Abstract
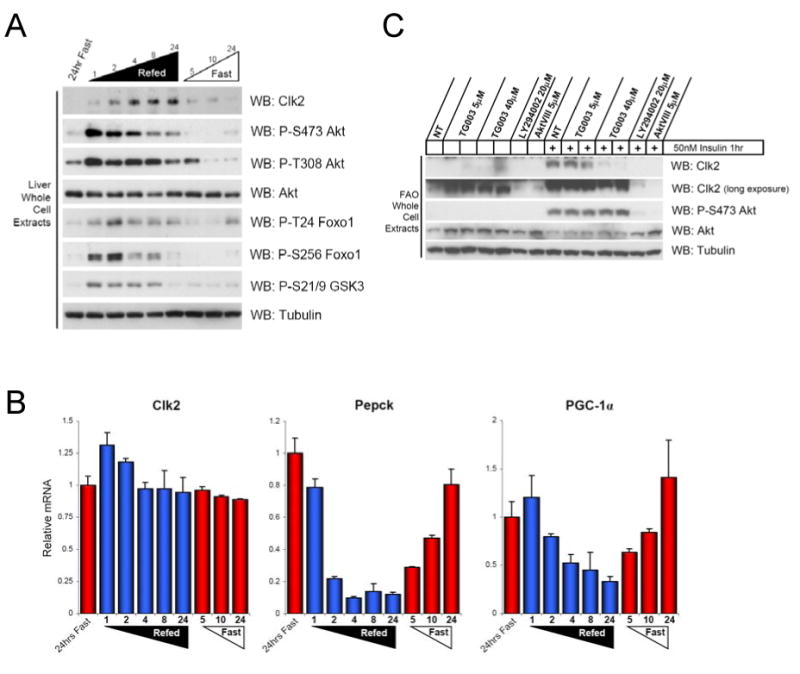
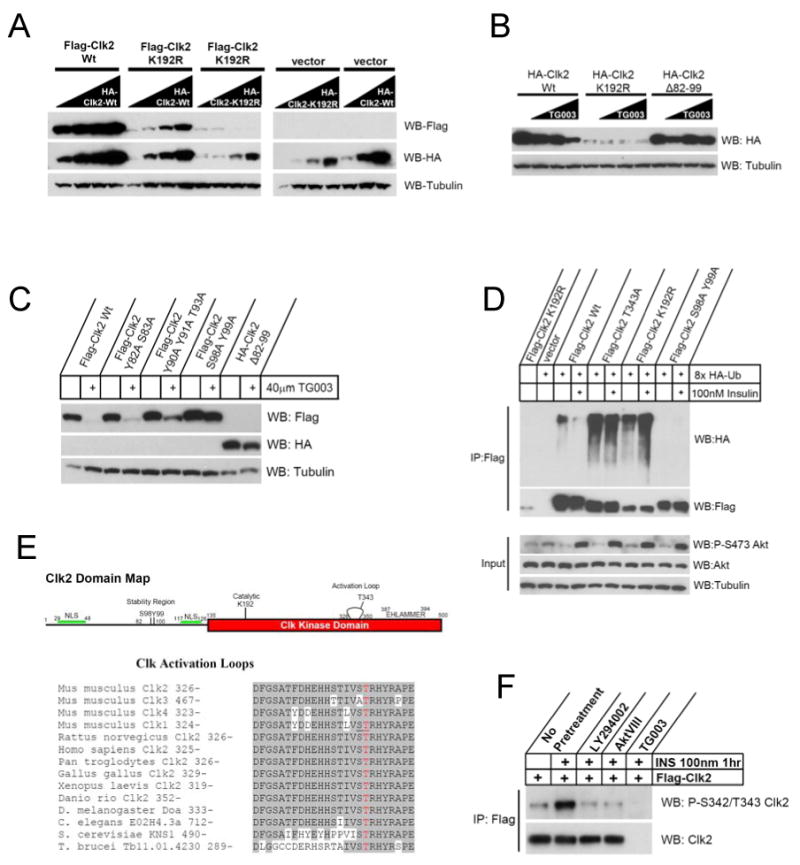
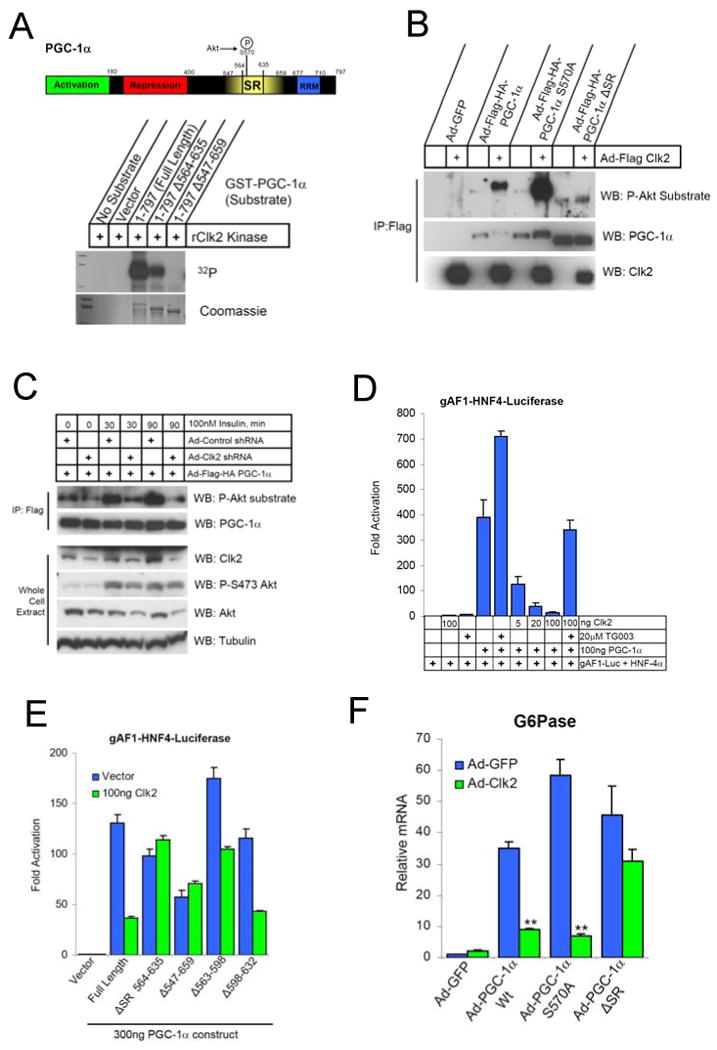
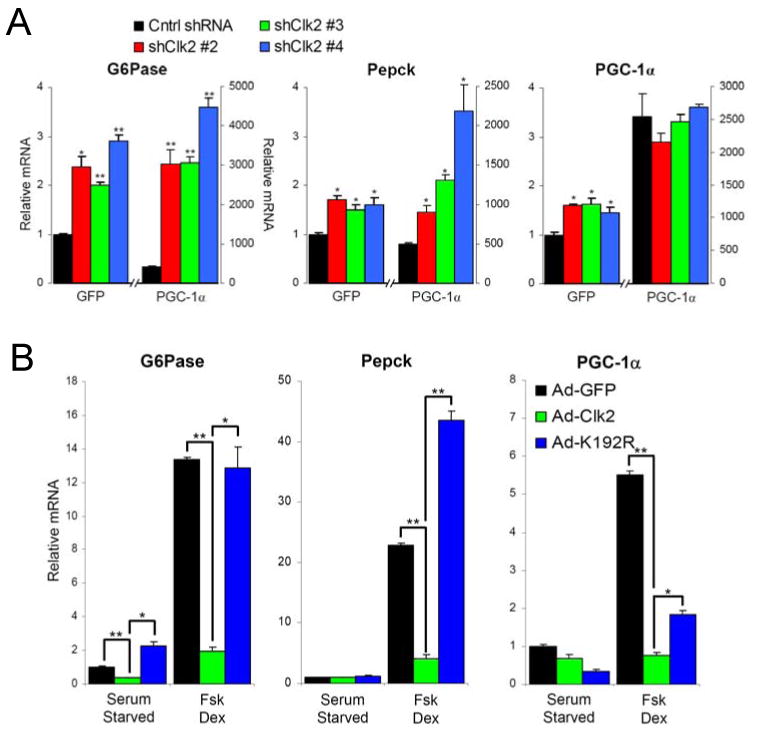
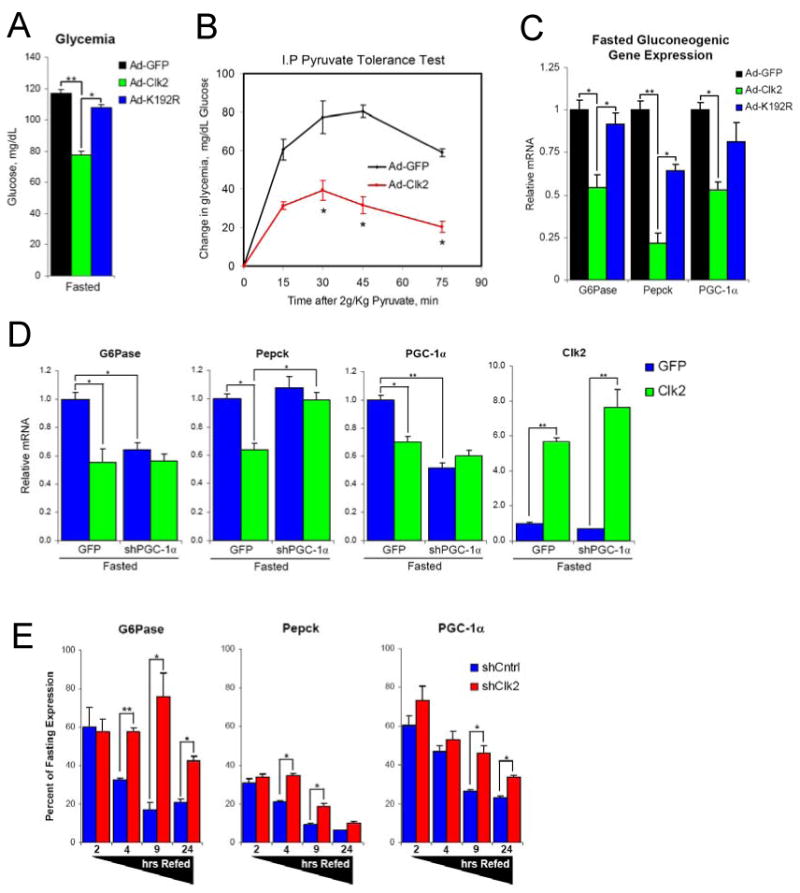
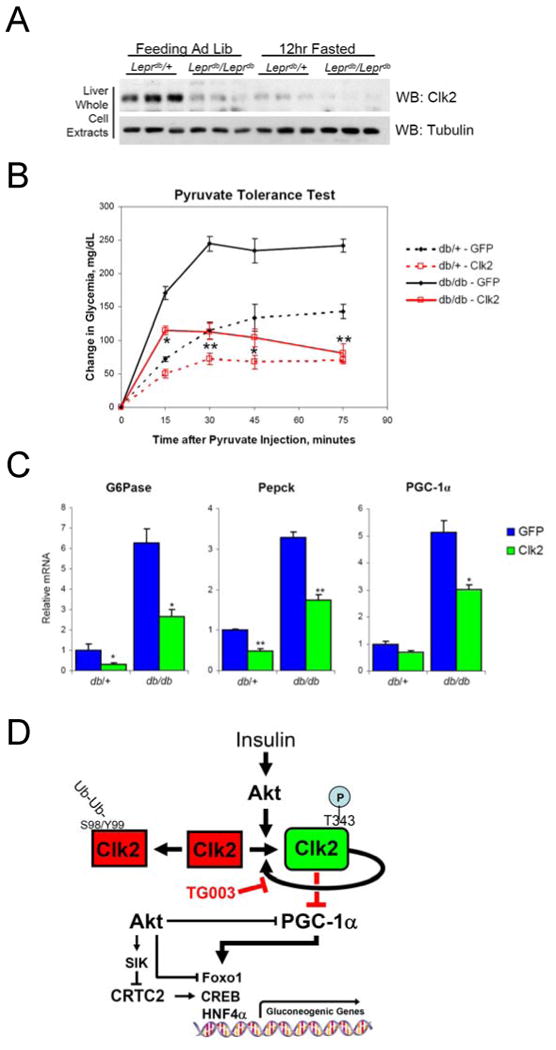
Dynamic regulation of insulin signaling and metabolic gene expression is critical to nutrient homeostasis; dysregulation of these pathways is widely implicated in insulin resistance and other disease states. Though the metabolic effects of insulin are well established, the components linking insulin signal transduction to a metabolic response are not as well understood. Here, we show that Cdc2-like kinase 2 (Clk2) is an insulin-regulated suppressor of hepatic gluconeogenesis and glucose output. Clk2 protein levels and kinase activity are induced as part of the hepatic refeeding response by the insulin/Akt pathway. Clk2 directly phosphorylates the SR domain on PGC-1alpha, resulting in repression of gluconeogenic gene expression and hepatic glucose output. In addition, Clk2 is downregulated in db/db mice, and reintroduction of Clk2 largely corrects glycemia. Thus, we have identified a role for and regulation of the Clk2 kinase as a component of hepatic insulin signaling and glucose metabolism.
2010 Elsevier Inc.
Figures
Comment in
-
Clking on PGC-1alpha to inhibit gluconeogenesis.Cell Metab. 2010 Jan;11(1):6-7. doi: 10.1016/j.cmet.2009.12.003. Cell Metab. 2010. PMID: 20085730 Free PMC article.
References
-
- Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annual review of physiology. 2006;68:123–158. - PubMed
-
- Colwill K, Feng LL, Yeakley JM, Gish GD, Caceres JF, Pawson T, Fu XD. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. The Journal of biological chemistry. 1996a;271:24569–24575. - PubMed
-
- Dentin R, Liu Y, Koo SH, Hedrick S, Vargas T, Heredia J, Yates J, 3rd, Montminy M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–369. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
