

Is abnormal axonal transport a cause, a contributing factor or a consequence of the neuronal pathology in Alzheimer's disease?
- PMID: 20076770
- PMCID: PMC2805861
- DOI: 10.2217/fnl.09.54
Is abnormal axonal transport a cause, a contributing factor or a consequence of the neuronal pathology in Alzheimer's disease?
Abstract
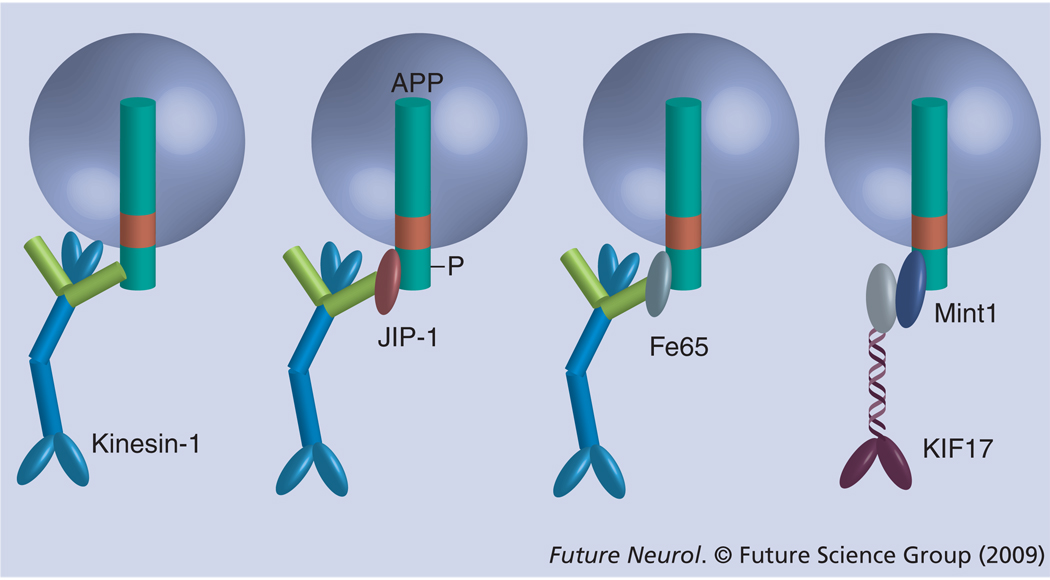
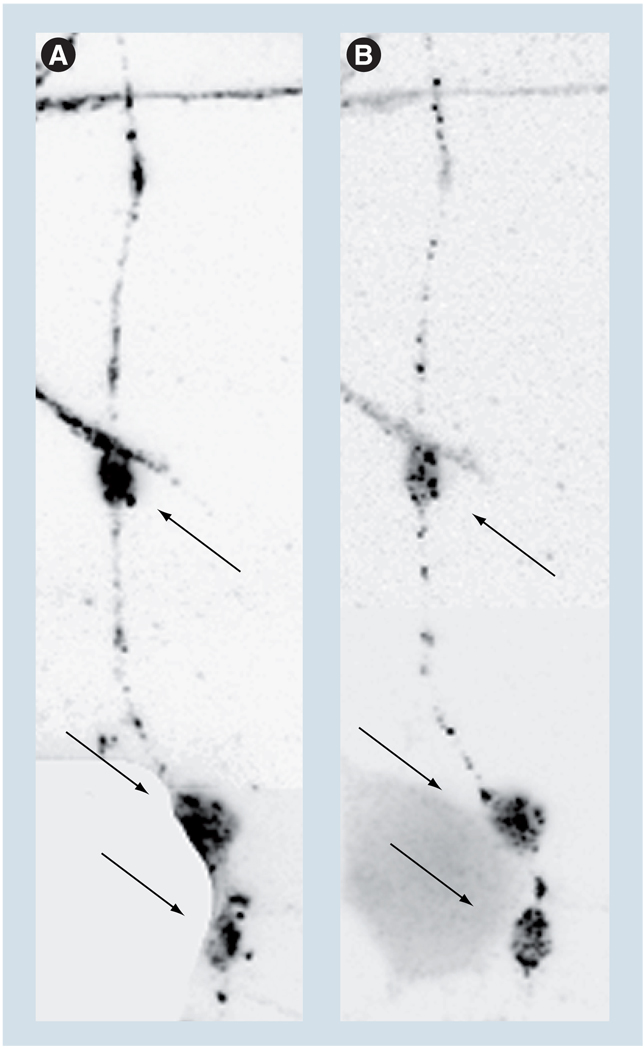
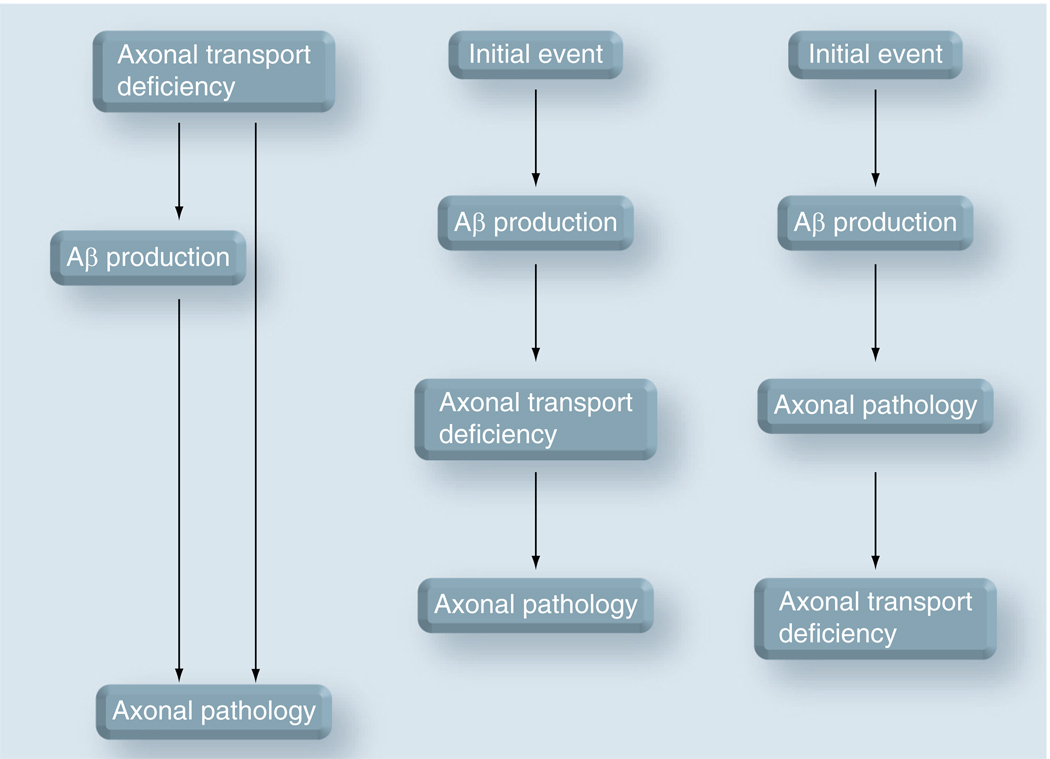
Axonal transport, the process by which membrane-bound organelles and soluble protein complexes are transported into and out of axons, ensures proper function of the neuron, including that of the synapse. As such, abnormalities in axonal transport could lead to neuronal pathology and disease. Similar to many neurodegenerative diseases, axonal transport is deficient in Alzheimer's disease (AD), a neurodegenerative brain disorder that affects old-age humans and is characterized by the deterioration of cognitive function and progressive memory loss. It was proposed that the synaptic pathology and neuronal degeneration that develops in AD could be caused by an abnormal axonal transport, and that the mutated proteins that cause early-onset AD, as well as the genetic variants that confer predisposition to late-onset AD might somehow impede axonal transport. This paper analyzes the data that support or contradict this hypothesis. Together, they indicate that, although abnormalities in axonal transport are part of the disease, additional studies are required to clearly establish to what extent deficient axonal transport is the cause or the effect of the neuronal pathology in AD, and to identify mechanisms that lead to its perturbation.
Figures


References
-
- Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. - PubMed
-
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. - PubMed
-
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999;399:A23–A31. - PubMed
-
- Hedera P, Turner RS. Inherited dementias. Neurol. Clin. 2002;20:779–808. vii. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials