

Clinical Diversity of SCN4A-Mutation-Associated Skeletal Muscle Sodium Channelopathy
- PMID: 20076800
- PMCID: PMC2806541
- DOI: 10.3988/jcn.2009.5.4.186
Clinical Diversity of SCN4A-Mutation-Associated Skeletal Muscle Sodium Channelopathy
Abstract
Background and purpose: Mutations of the skeletal muscle sodium channel gene SCN4A, which is located on chromosome 17q23-25, are associated with various neuromuscular disorders that are labeled collectively as skeletal muscle sodium channelopathy. These disorders include hyperkalemic periodic paralysis (HYPP), hypokalemic periodic paralysis, paramyotonia congenita (PMC), potassium-aggravated myotonia, and congenital myasthenic syndrome. This study analyzed the clinical and mutational spectra of skeletal muscle sodium channelopathy in Korean subjects.
Methods: Six unrelated Korean patients with periodic paralysis or nondystrophic myotonia associated with SCN4A mutations were included in the study. For the mutational analysis of SCN4A, we performed a full sequence analysis of the gene using the patients' DNA. We also analyzed the patients' clinical history, physical findings, laboratory tests, and responses to treatment.
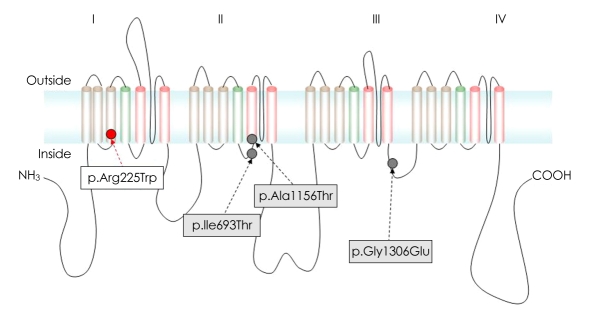
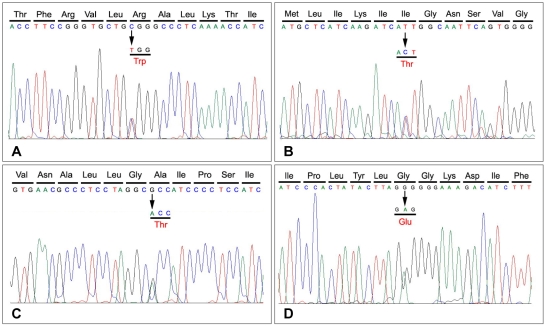
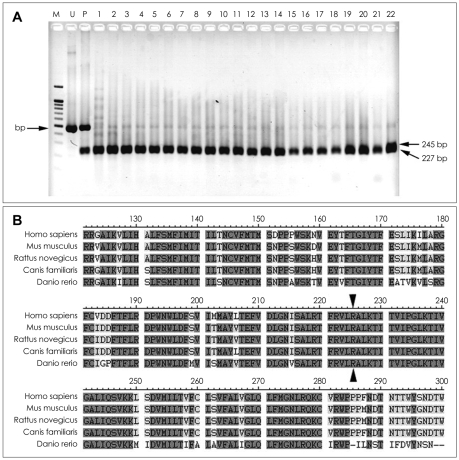
Results: We identified four different mutations (one of which was novel) in all of the patients examined. The novel heterozygous missense mutation, p.R225W, was found in one patient with mild nonpainful myotonia. Our patients exhibited various clinical phenotypes: pure myotonia in four, and PMC in one, and HYPP in one. The four patients with pure myotonia were initially diagnosed as having myotonia congenita (MC), but a previous analysis revealed no CLCN1 mutation.
Conclusions: Clinical differentiating between sodium-channel myotonia (SCM) and MC is not easy, and it is suggested that a mutational analysis of both SCN4A and CLCN1 is essential for the differential diagnosis of SCM and MC.
Keywords: SCN4A; Wordsaamyotonic disorders; familial periodic paralyses.
Figures
References
-
- George AL, Jr, Ledbetter DH, Kallen RG, Barchi RL. Assignment of a human skeletal muscle sodium channel alpha-subunit gene (SCN4A) to 17q23.1-25.3. Genomics. 1991;9:555–556. - PubMed
-
- George AL, Jr, Crackower MA, Abdalla JA, Hudson AJ, Ebers GC. Molecular basis of Thomsen's disease (autosomal dominant myotonia congenita) Nat Genet. 1993;3:305–310. - PubMed
-
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. - PubMed
-
- Ptácek LJ, George AL, Jr, Griggs RC, Tawil R, Kallen RG, Barchi RL, et al. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell. 1991;67:1021–1027. - PubMed
-
- McClatchey AI, McKenna-Yasek D, Cros D, Worthen HG, Kuncl RW, DeSilva SM, et al. Novel mutations in families with unusual and variable disorders of the skeletal muscle sodium channel. Nat Genet. 1992;2:148–152. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
