

Mitochondrial energetics and therapeutics
- PMID: 20078222
- PMCID: PMC3245719
- DOI: 10.1146/annurev.pathol.4.110807.092314
Mitochondrial energetics and therapeutics
Abstract
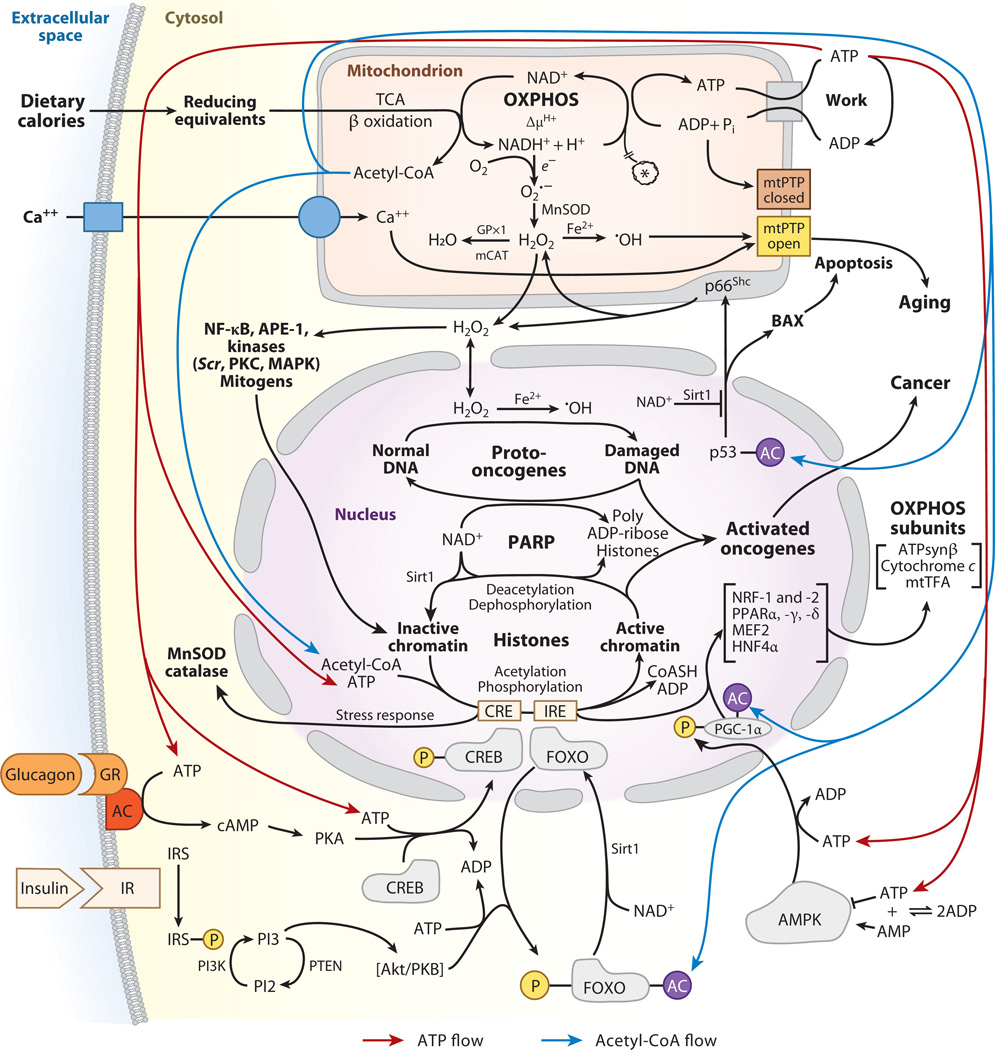
Mitochondrial dysfunction has been linked to a wide range of degenerative and metabolic diseases, cancer, and aging. All these clinical manifestations arise from the central role of bioenergetics in cell biology. Although genetic therapies are maturing as the rules of bioenergetic genetics are clarified, metabolic therapies have been ineffectual. This failure results from our limited appreciation of the role of bioenergetics as the interface between the environment and the cell. A systems approach, which, ironically, was first successfully applied over 80 years ago with the introduction of the ketogenic diet, is required. Analysis of the many ways that a shift from carbohydrate glycolytic metabolism to fatty acid and ketone oxidative metabolism may modulate metabolism, signal transduction pathways, and the epigenome gives us an appreciation of the ketogenic diet and the potential for bioenergetic therapeutics.
Figures


References
-
- Wallace DC, Lott MT, Procaccio V. Mitochondrial genes in degenerative diseases, cancer and aging. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Emery and Rimoin’s Principles and Practice of Medical Genetics. 5th ed. Vol. 1. Philadelphia: Churchill Livingstone Elsevier; 2007. pp. 194–298.
-
- Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. - PubMed
-
- Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
