

Halogenated imidazole derivatives block RNA polymerase II elongation along mitogen inducible genes
- PMID: 20078881
- PMCID: PMC2824761
- DOI: 10.1186/1471-2199-11-4
Halogenated imidazole derivatives block RNA polymerase II elongation along mitogen inducible genes
Abstract
Background: Aberrant activation of protein kinases is one of the essential oncogenic driving forces inherent to the process of tumorigenesis. The protein kinase CK2 plays an important role in diverse biological processes, including cell growth and proliferation as well as in the governing and transduction of prosurvival signals. Increased expression of CK2 is a hallmark of some cancers, hence its antiapoptotic properties may be relevant to cancer onset. Thus, the designing and synthesis of the CK2 inhibitors has become an important pursuit in the search for cancer therapies.
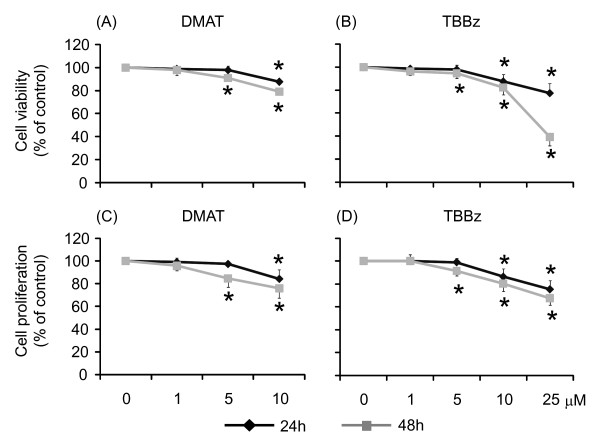
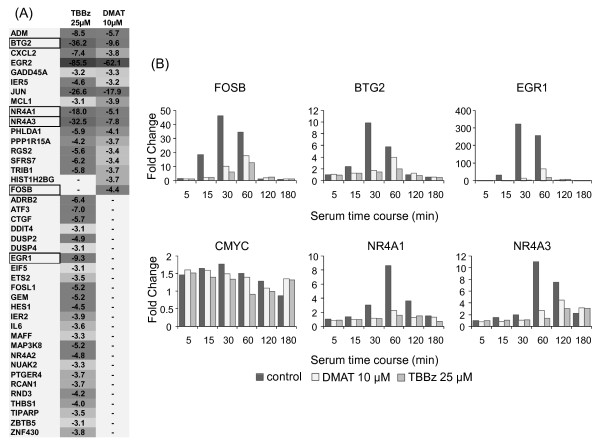
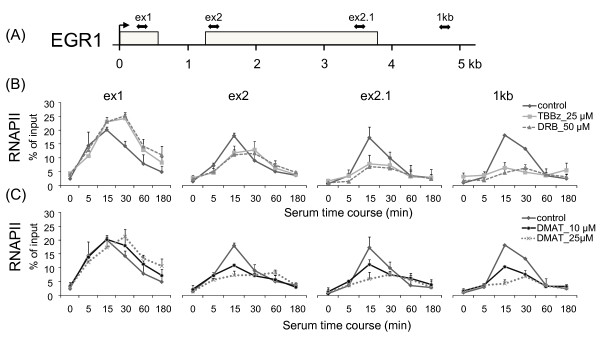
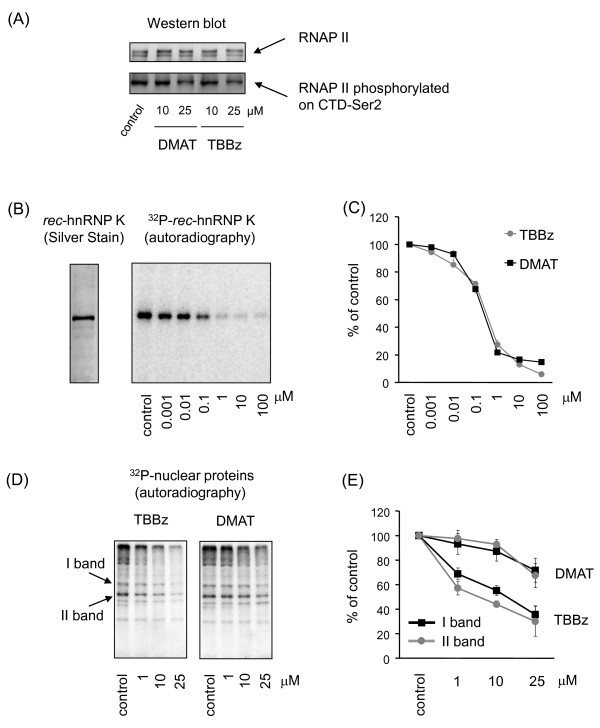
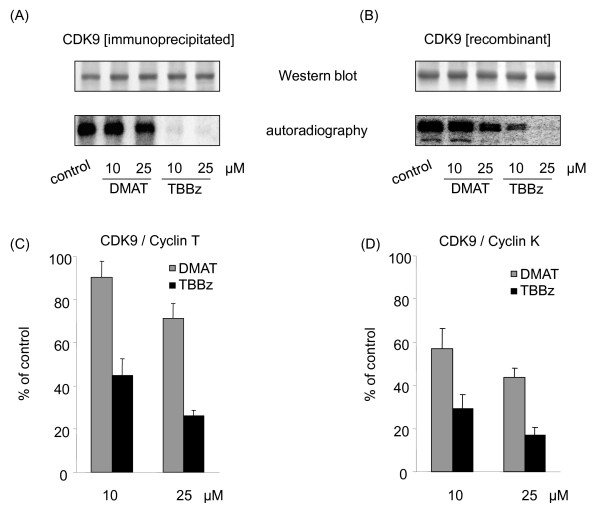
Results: Using a high-throughput microarray approach, we demonstrate that two potent inhibitors of CK2, 4,5,6,7-tetrabromo-benzimidazole (TBBz) and 2-Dimethyloamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT), blocked mitogen induced mRNA expression of immediate early genes. Given the impact of these inhibitors on the process of transcription, we investigated their effects on RNA Polymerase II (RNAPII) elongation along the mitogen inducible gene, EGR1 (early growth response 1), using chromatin immunoprecipitation (ChIP) assay. ChIP analysis demonstrated that both drugs arrest RNAPII elongation. Finally, we show that CDK9 kinase activity, essential for the triggering of RNAPII elongation, was blocked by TBBz and to lesser degree by DMAT.
Conclusions: Our approach revealed that small molecules derived from halogenated imidazole compounds may decrease cell proliferation, in part, by inhibiting pathways that regulate transcription elongation.
Figures
References
-
- Duncan JS, Litchfield DW. Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochimica et biophysica acta. 2008;1784(1):33–47. - PubMed
-
- Kramerov AA, Saghizadeh M, Caballero S, Shaw LC, Li Calzi S, Bretner M, Montenarh M, Pinna LA, Grant MB, Ljubimov AV. Inhibition of protein kinase CK2 suppresses angiogenesis and hematopoietic stem cell recruitment to retinal neovascularization sites. Molecular and cellular biochemistry. 2008;316(1-2):177–186. doi: 10.1007/s11010-008-9831-4. - DOI - PMC - PubMed
-
- Perea SE, Reyes O, Baladron I, Perera Y, Farina H, Gil J, Rodriguez A, Bacardi D, Marcelo JL, Cosme K. CIGB-300, a novel proapoptotic peptide that impairs the CK2 phosphorylation and exhibits anticancer properties both in vitro and in vivo. Molecular and cellular biochemistry. 2008;316(1-2):163–167. doi: 10.1007/s11010-008-9814-5. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
