

Interactions between PTB RRMs induce slow motions and increase RNA binding affinity
- PMID: 20080103
- PMCID: PMC2830284
- DOI: 10.1016/j.jmb.2009.12.051
Interactions between PTB RRMs induce slow motions and increase RNA binding affinity
Abstract
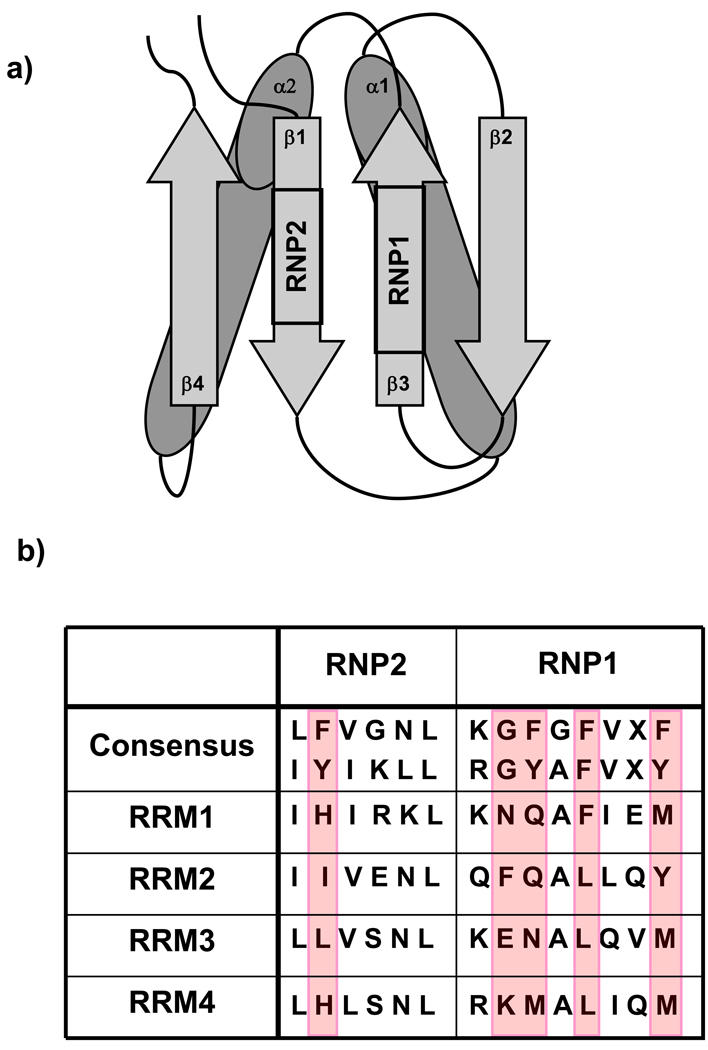
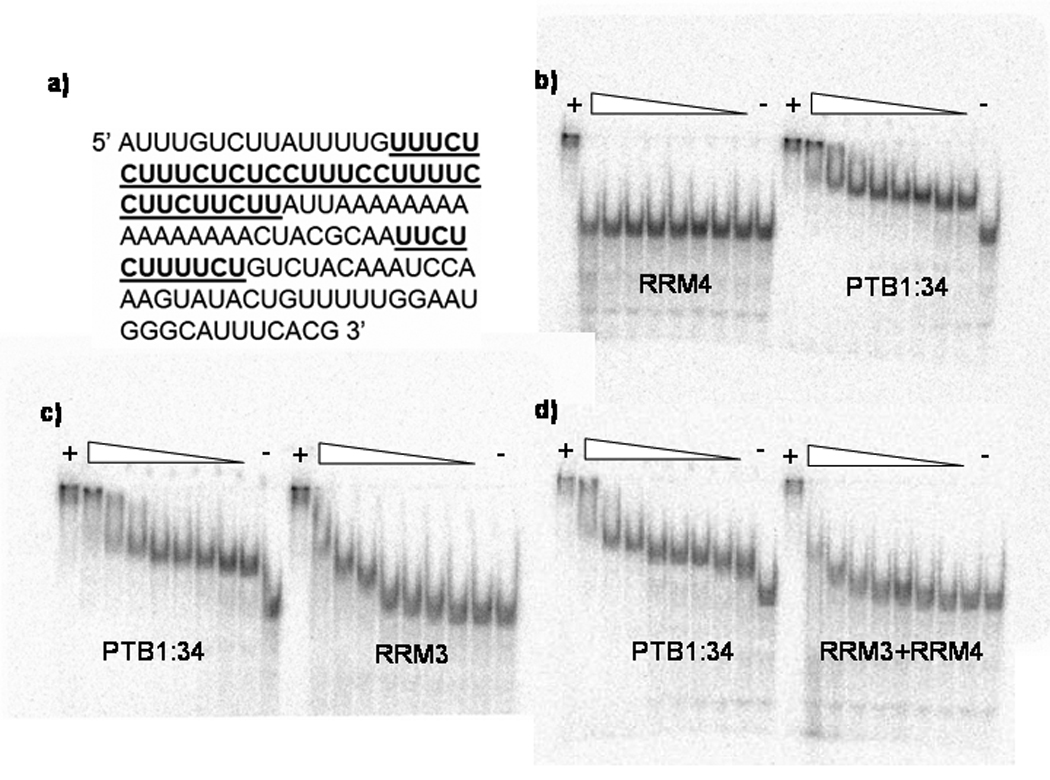
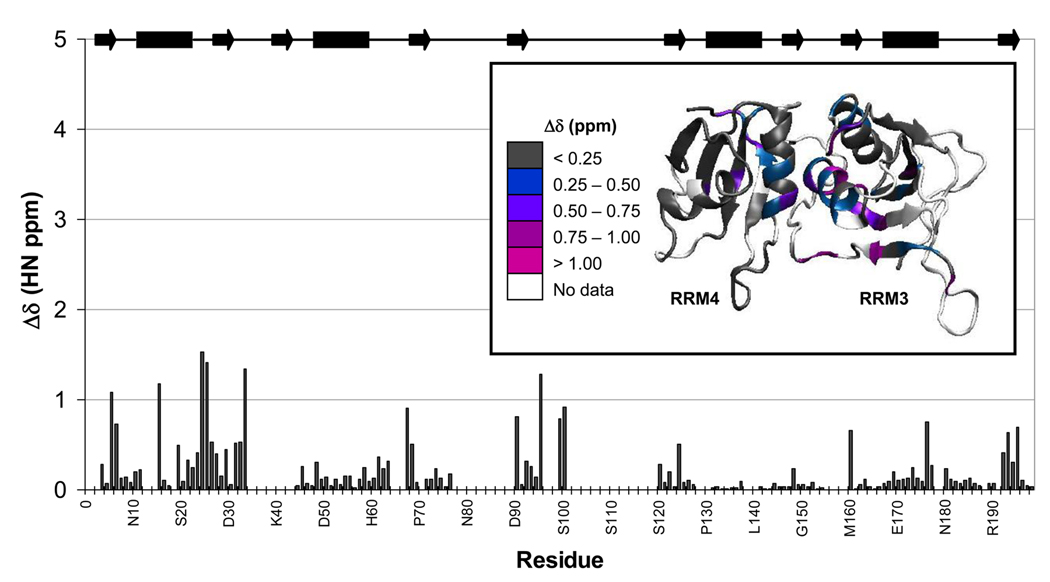
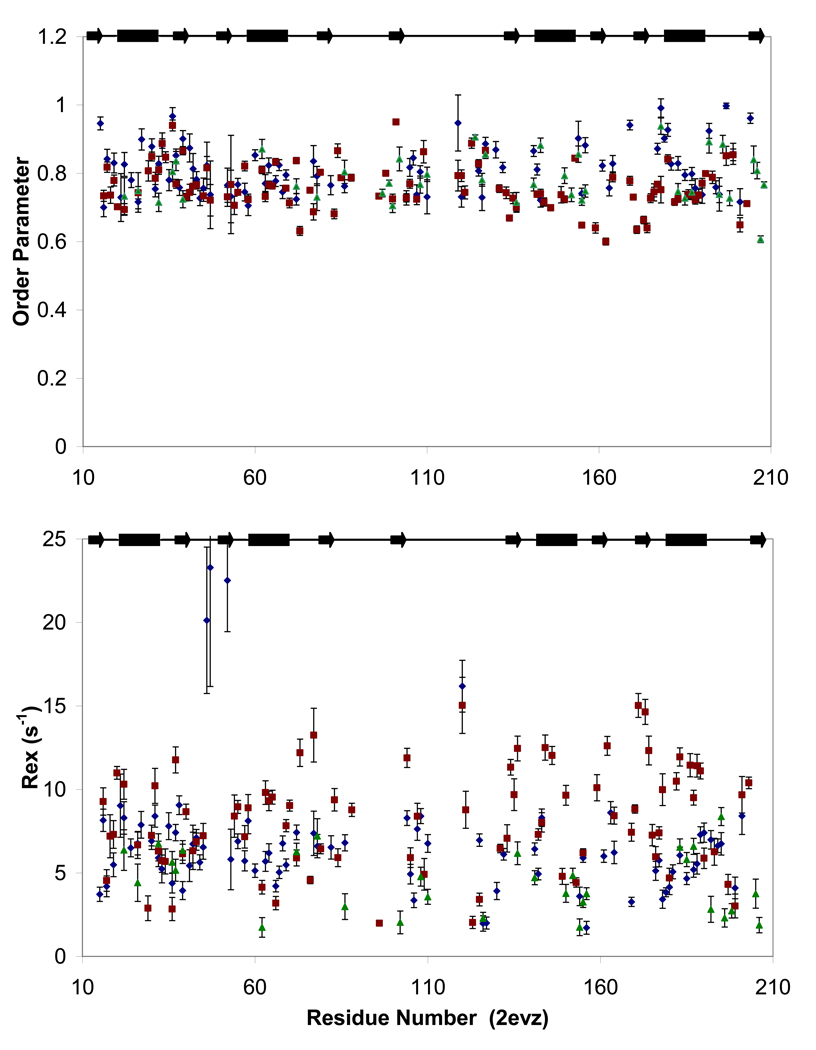
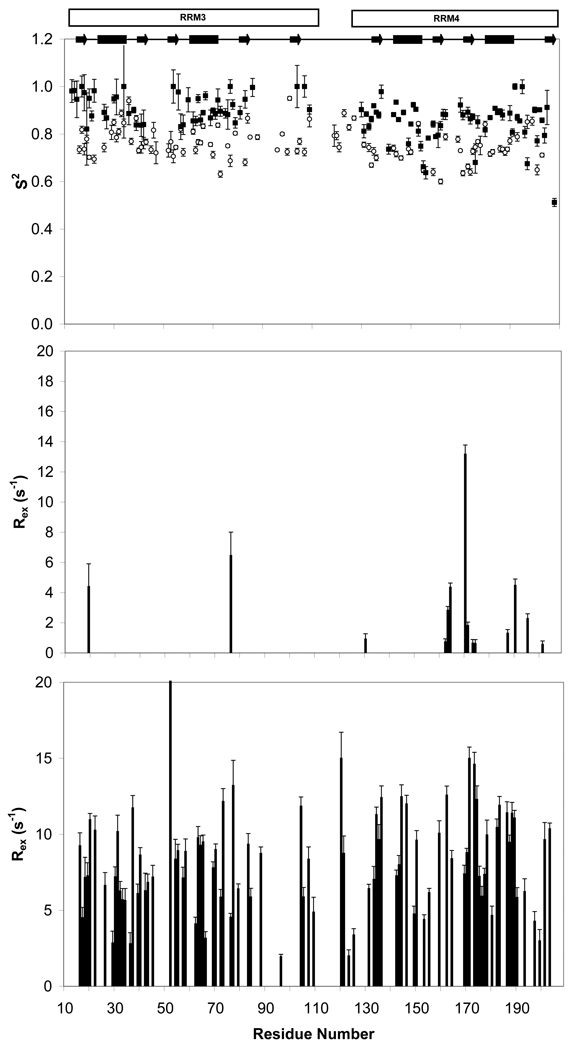
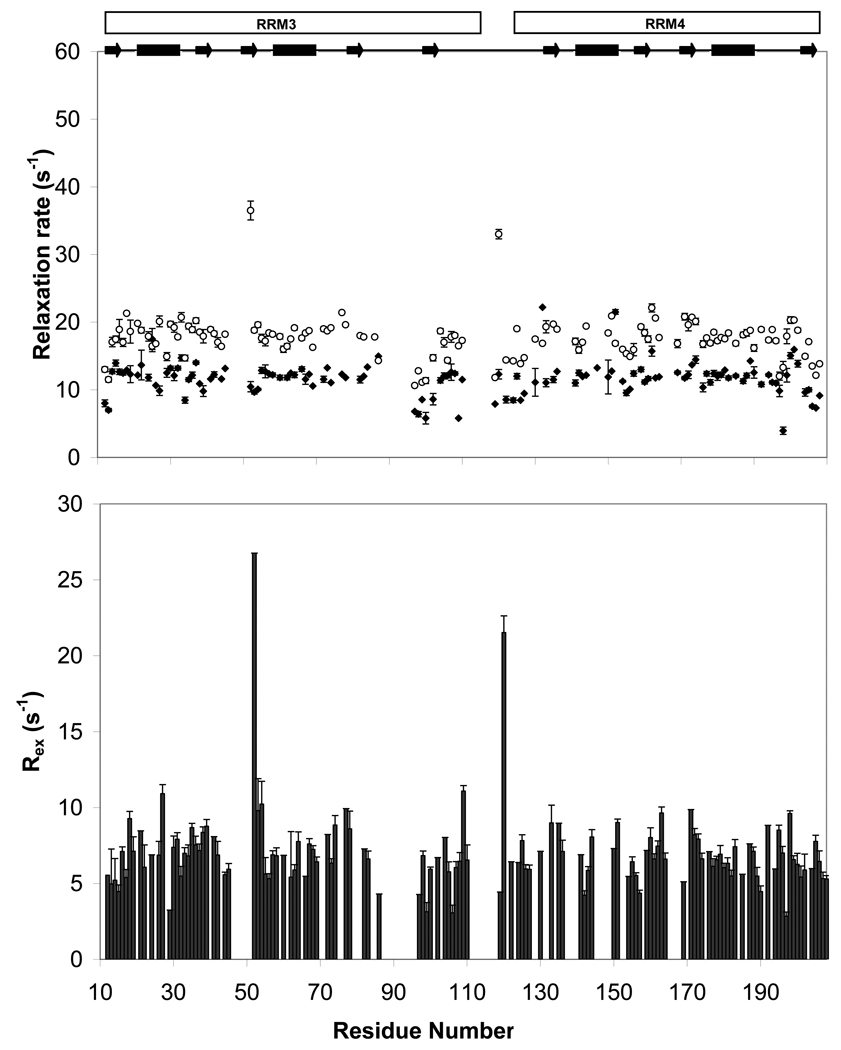
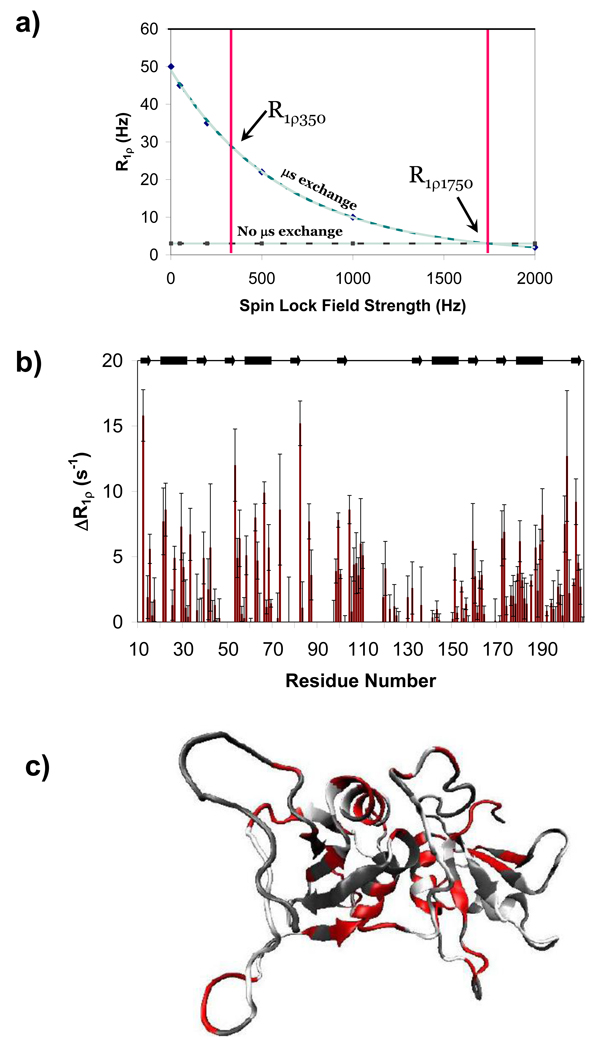
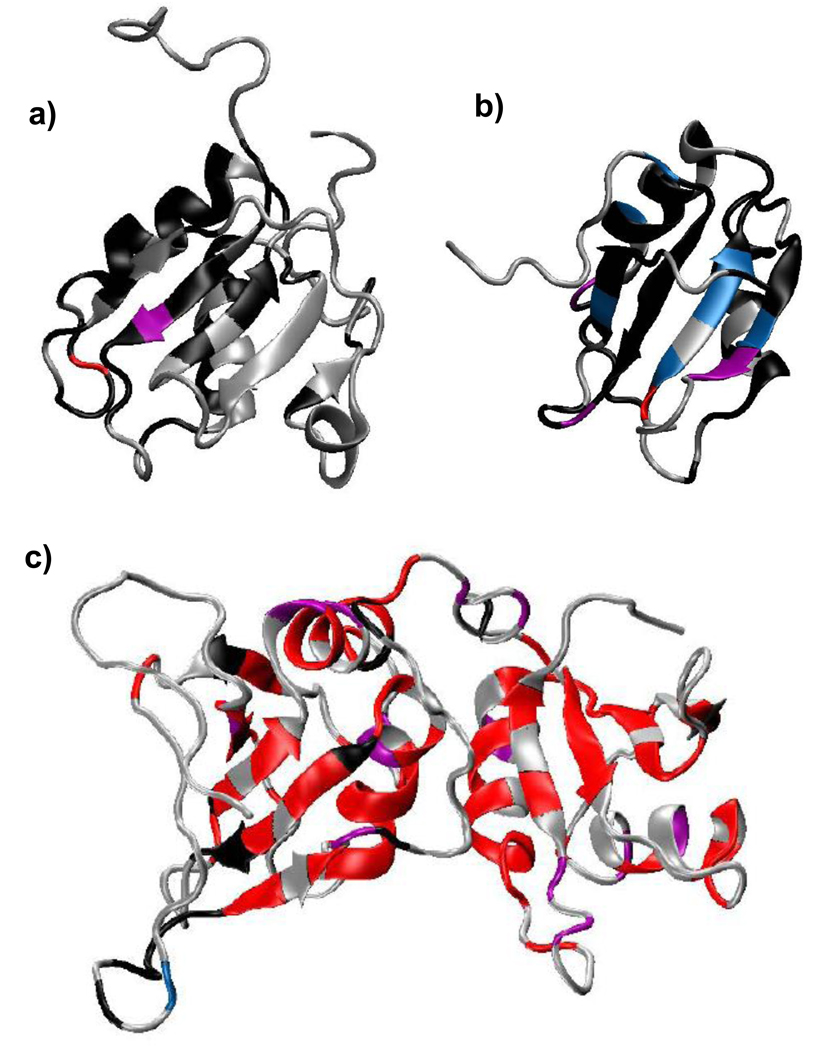
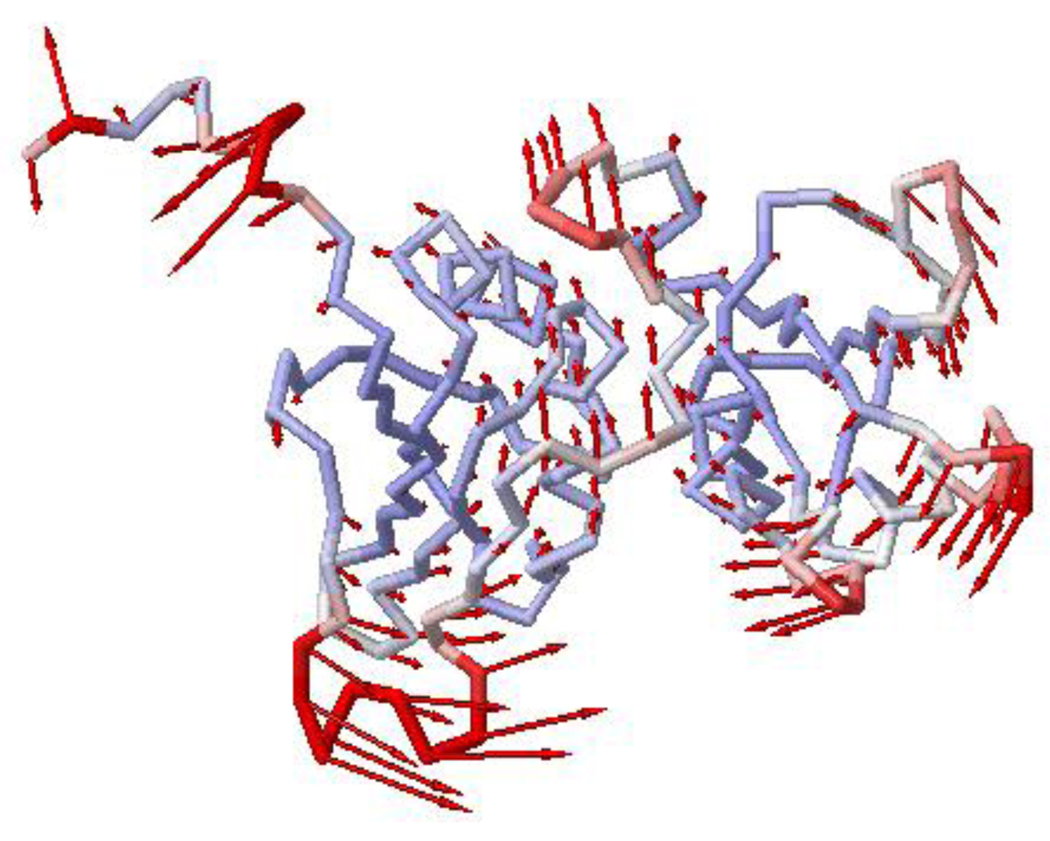
Polypyrimidine tract binding protein (PTB) participates in a variety of functions in eukaryotic cells, including alternative splicing, mRNA stabilization, and internal ribosomal entry site-mediated translation initiation. Its mechanism of RNA recognition is determined in part by the novel geometry of its two C-terminal RNA recognition motifs (RRM3 and RRM4), which interact with each other to form a stable complex (PTB1:34). This complex itself is unusual among RRMs, suggesting that it performs a specific function for the protein. In order to understand the advantage it provides to PTB, the fundamental properties of PTB1:34 are examined here as a comparative study of the complex and its two constituent RRMs. Both RRM3 and RRM4 adopt folded structures that NMR data show to be similar to their structure in PRB1:34. The RNA binding properties of the domains differ dramatically. The affinity of each separate RRM for polypyrimidine tracts is far weaker than that of PTB1:34, and simply mixing the two RRMs does not create an equivalent binding platform. (15)N NMR relaxation experiments show that PTB1:34 has slow, microsecond motions throughout both RRMs including the interdomain linker. This is in contrast to the individual domains, RRM3 and RRM4, where only a few backbone amides are flexible on this time scale. The slow backbone dynamics of PTB1:34, induced by packing of RRM3 and RRM4, could be essential for high-affinity binding to a flexible polypyrimidine tract RNA and also provide entropic compensation for its own formation.
Copyright (c) 2009 Elsevier Ltd. All rights reserved.
Figures

Similar articles
-
Structure and RNA interactions of the N-terminal RRM domains of PTB.Structure. 2004 Sep;12(9):1631-43. doi: 10.1016/j.str.2004.07.008. Structure. 2004. PMID: 15341728
-
Polypyrimidine-Tract-Binding Protein Isoforms Differentially Regulate the Hepatitis C Virus Internal Ribosome Entry Site.Viruses. 2022 Dec 20;15(1):8. doi: 10.3390/v15010008. Viruses. 2022. PMID: 36680049 Free PMC article.
-
Characterization of multimeric complexes formed by the human PTB1 protein on RNA.RNA. 2006 Mar;12(3):457-75. doi: 10.1261/rna.2178406. Epub 2006 Jan 23. RNA. 2006. PMID: 16431980 Free PMC article.
-
Structure-function relationships of the polypyrimidine tract binding protein.Cell Mol Life Sci. 2008 Feb;65(4):516-27. doi: 10.1007/s00018-007-7378-2. Cell Mol Life Sci. 2008. PMID: 17975705 Free PMC article. Review.
-
Defining the roles and interactions of PTB.Biochem Soc Trans. 2012 Aug;40(4):815-20. doi: 10.1042/BST20120044. Biochem Soc Trans. 2012. PMID: 22817740 Review.
Cited by
-
The organization of RNA contacts by PTB for regulation of FAS splicing.Nucleic Acids Res. 2014 Jul;42(13):8605-20. doi: 10.1093/nar/gku519. Epub 2014 Jun 23. Nucleic Acids Res. 2014. PMID: 24957602 Free PMC article.
-
Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2.Crit Rev Biochem Mol Biol. 2012 Jul-Aug;47(4):360-78. doi: 10.3109/10409238.2012.691456. Epub 2012 Jun 2. Crit Rev Biochem Mol Biol. 2012. PMID: 22655688 Free PMC article.
-
RNA and Proteins: Mutual Respect.F1000Res. 2017 Mar 27;6:345. doi: 10.12688/f1000research.10572.1. eCollection 2017. F1000Res. 2017. PMID: 28408981 Free PMC article. Review.
-
Solution and crystal structures of a C-terminal fragment of the neuronal isoform of the polypyrimidine tract binding protein (nPTB).PeerJ. 2014 Mar 13;2:e305. doi: 10.7717/peerj.305. eCollection 2014. PeerJ. 2014. PMID: 24688880 Free PMC article.
-
Intrinsic dynamics of an extended hydrophobic core in the S. cerevisiae RNase III dsRBD contributes to recognition of specific RNA binding sites.J Mol Biol. 2013 Feb 8;425(3):546-62. doi: 10.1016/j.jmb.2012.11.025. Epub 2012 Nov 28. J Mol Biol. 2013. PMID: 23201338 Free PMC article.
References
-
- Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. - PubMed
-
- Eisenmesser EZ, et al. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. - PubMed
-
- Bracken C, Carr PA, Cavanagh J, Palmer AG. Temperature dependence of intramolecular dynamics of the basic leucine zipper of GCN4: Implications for the entropy of association with DNA. J. Mol. Biol. 1999;285:2133–2146. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
