

Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65
- PMID: 20080798
- PMCID: PMC2806709
- DOI: 10.1073/pnas.0912493107
Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65
Abstract
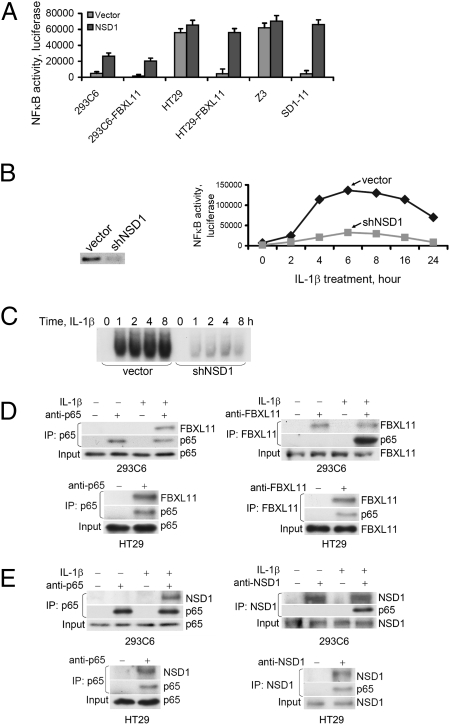
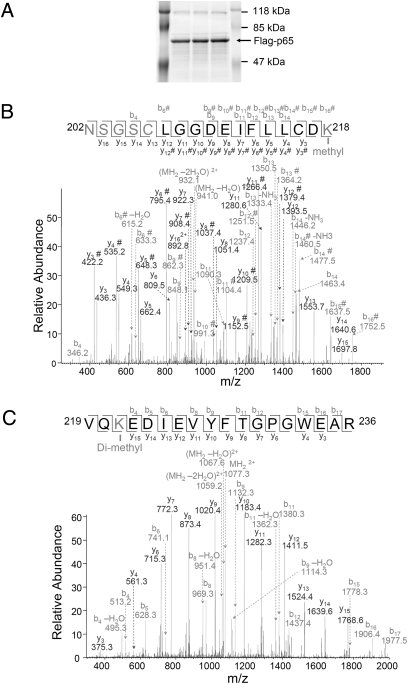
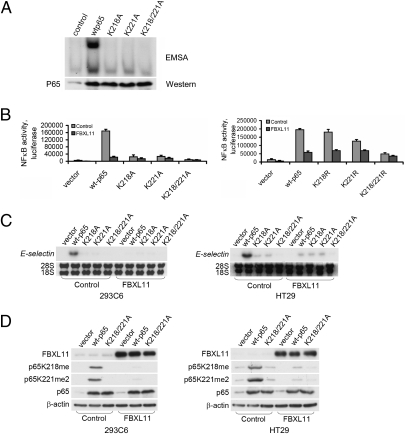
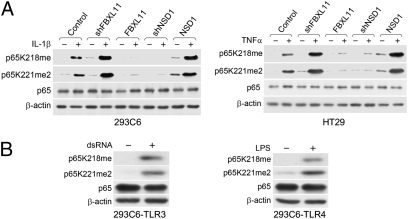
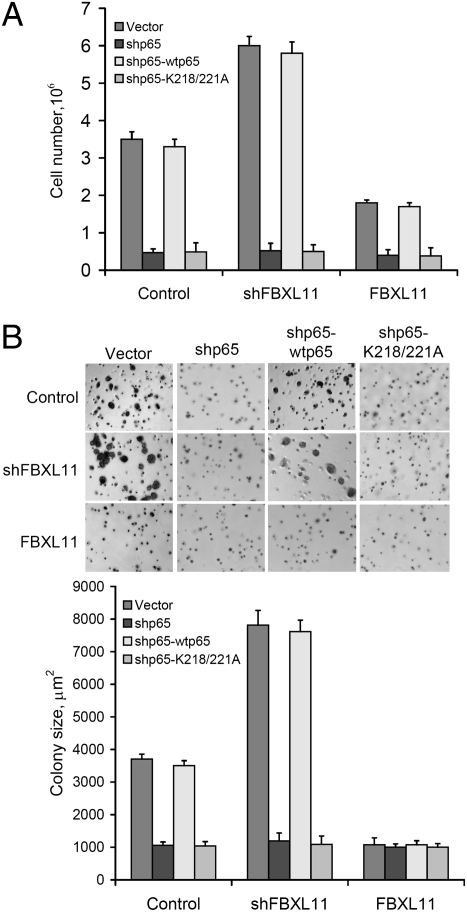
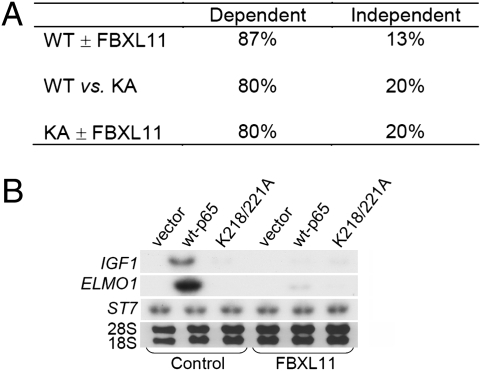
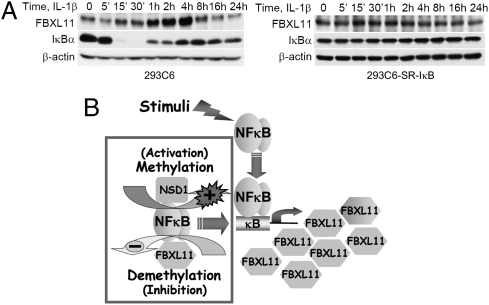
NF-kappaB, a central coordinator of immune and inflammatory responses, must be tightly regulated. We describe a NF-kappaB regulatory pathway that is driven by reversible lysine methylation of the p65 subunit, carried out by a lysine methylase, the nuclear receptor-binding SET domain-containing protein 1 (NSD1), and a lysine demethylase, F-box and leucine-rich repeat protein 11 (FBXL11). Overexpression of FBXL11 inhibits NF-kappaB activity, and a high level of NSD1 activates NF-kappaB and reverses the inhibitory effect of FBXL11, whereas reduced expression of NSD1 decreases NF-kappaB activation. The targets are K218 and K221 of p65, which are methylated in cells with activated NF-kappaB. Overexpression of FBXL11 slowed the growth of HT29 cancer cells, whereas shRNA-mediated knockdown had the opposite effect, and these phenotypes were dependent on K218/K221 methylation. In mouse embryo fibroblasts, the activation of most p65-dependent genes relied on K218/K221 methylation. Importantly, expression of the FBXL11 gene is driven by NF-kappaB, revealing a negative regulatory feedback loop. We conclude that reversible lysine methylation of NF-kappaB is an important element in the complex regulation of this key transcription factor.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Karin M, Yamamoto Y, Wang QM. The IKK NF-κB system: A treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. - PubMed
-
- Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441:431–436. - PubMed
-
- Lu T, Sathe SS, Swiatkowski SM, Hampole CV, Stark GR. Secretion of cytokines and growth factors as a general cause of constitutive NFκB activation in cancer. Oncogene. 2004;23:2138–2145. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
