

Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer
- PMID: 20080953
- PMCID: PMC2807352
- DOI: 10.1101/gad.552910
Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer
Abstract
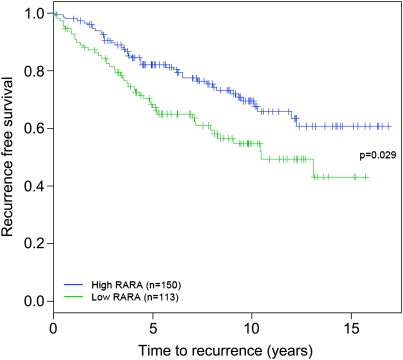
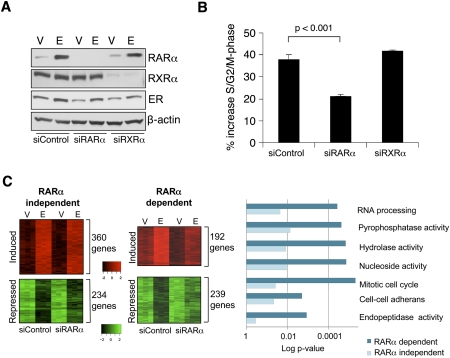
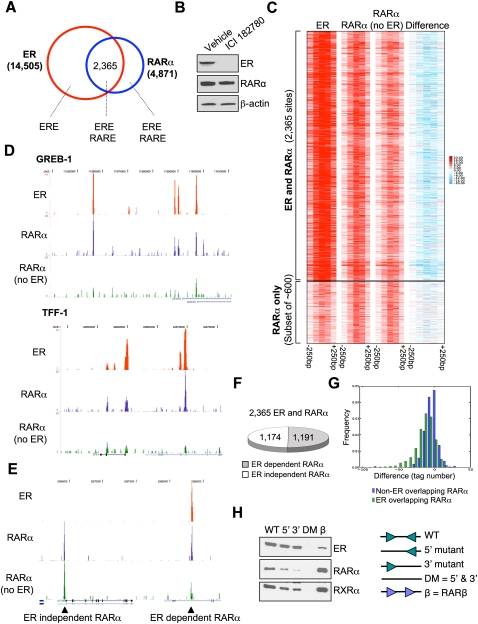
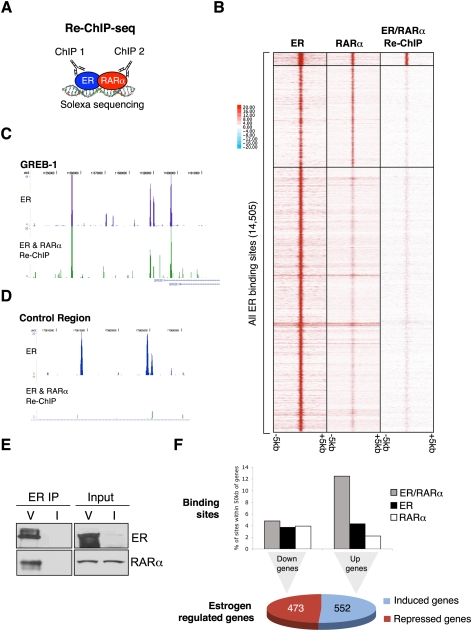
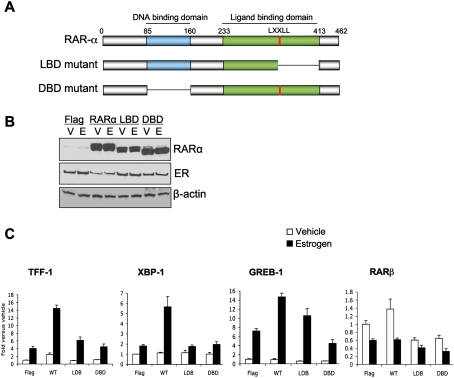
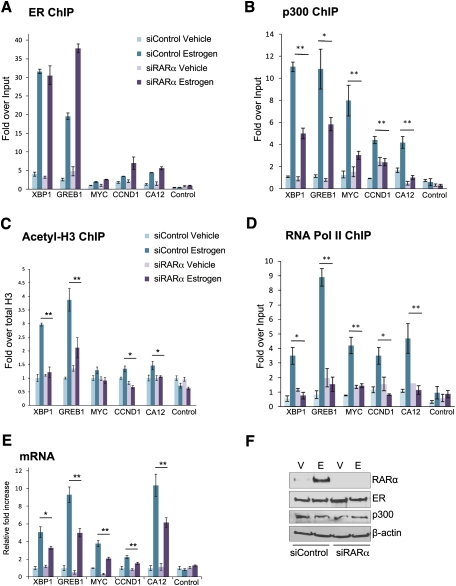
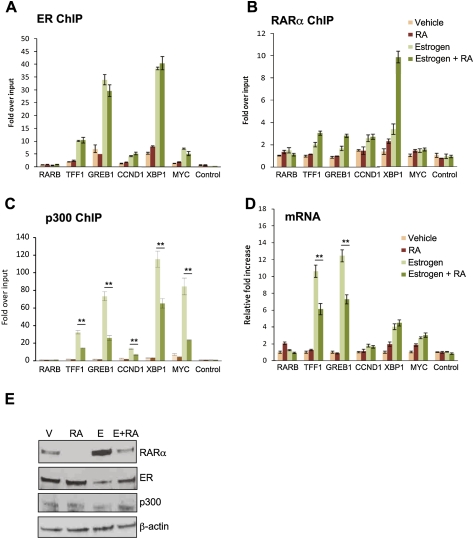
Retinoic acid receptor-alpha (RAR alpha) is a known estrogen target gene in breast cancer cells. The consequence of RAR alpha induction by estrogen was previously unknown. We now show that RAR alpha is required for efficient estrogen receptor-alpha (ER)-mediated transcription and cell proliferation. RAR alpha can interact with ER-binding sites, but this occurs in an ER-dependent manner, providing a novel role for RAR alpha that is independent of its classic role. We show, on a genome-wide scale, that RAR alpha and ER can co-occupy regulatory regions together within the chromatin. This transcriptionally active co-occupancy and dependency occurs when exposed to the predominant breast cancer hormone, estrogen--an interaction that is promoted by the estrogen-ER induction of RAR alpha. These findings implicate RAR alpha as an essential component of the ER complex, potentially by maintaining ER-cofactor interactions, and suggest that different nuclear receptors can cooperate for effective transcriptional activity in breast cancer cells.
Figures
References
-
- Balmer JE, Blomhoff R. A robust characterization of retinoic acid response elements based on a comparison of sites in three species. J Steroid Biochem Mol Biol. 2005;96:347–354. - PubMed
-
- Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 2004;328:1–16. - PubMed
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300.
-
- Borrow J, Goddard AD, Sheer D, Solomon E. Molecular analysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science. 1990;249:1577–1580. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases