

Protein secondary structure appears to be robust under in silico evolution while protein disorder appears not to be
- PMID: 20081223
- PMCID: PMC2828120
- DOI: 10.1093/bioinformatics/btq012
Protein secondary structure appears to be robust under in silico evolution while protein disorder appears not to be
Abstract
Motivation: The mutation of amino acids often impacts protein function and structure. Mutations without negative effect sustain evolutionary pressure. We study a particular aspect of structural robustness with respect to mutations: regular protein secondary structure and natively unstructured (intrinsically disordered) regions. Is the formation of regular secondary structure an intrinsic feature of amino acid sequences, or is it a feature that is lost upon mutation and is maintained by evolution against the odds? Similarly, is disorder an intrinsic sequence feature or is it difficult to maintain? To tackle these questions, we in silico mutated native protein sequences into random sequence-like ensembles and monitored the change in predicted secondary structure and disorder.
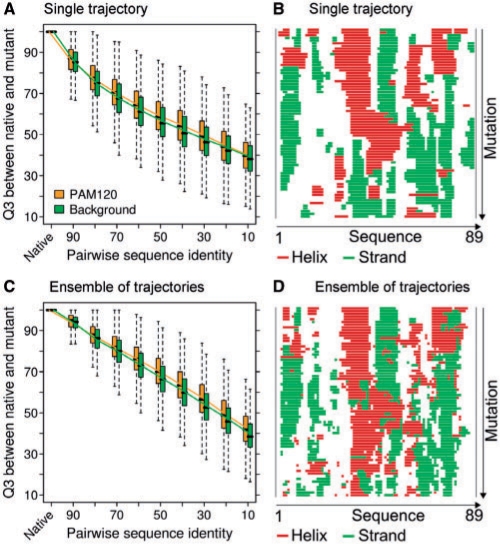
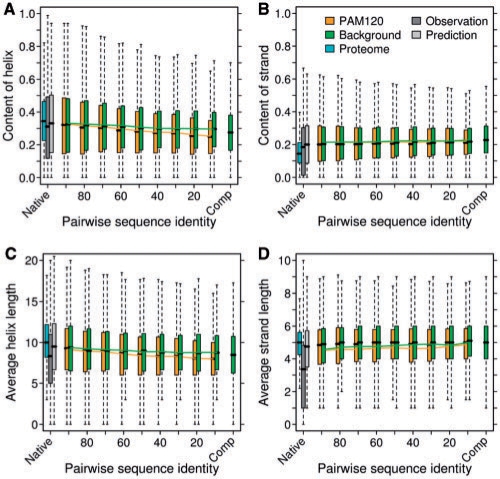
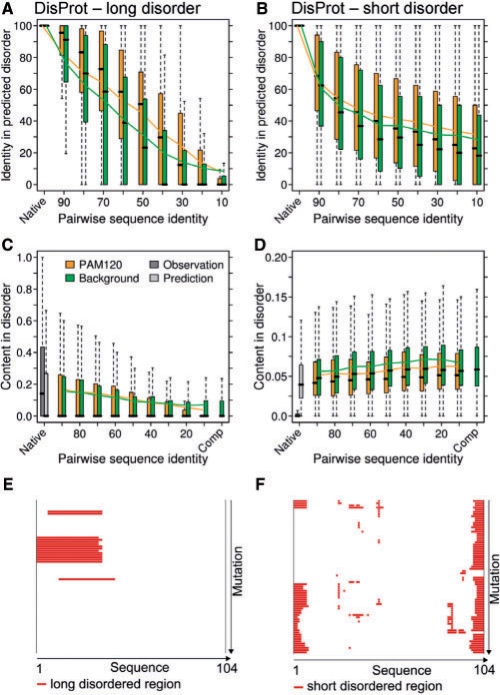
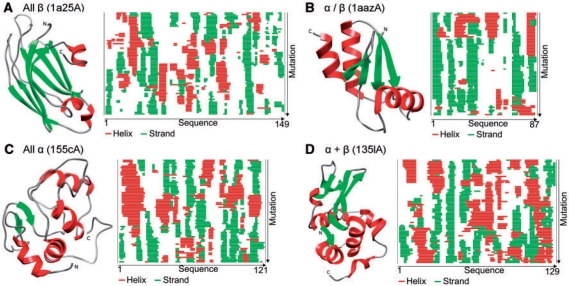
Results: We established that by our coarse-grained measures for change, predictions and observations were similar, suggesting that our results were not biased by prediction mistakes. Changes in secondary structure and disorder predictions were linearly proportional to the change in sequence. Surprisingly, neither the content nor the length distribution for the predicted secondary structure changed substantially. Regions with long disorder behaved differently in that significantly fewer such regions were predicted after a few mutation steps. Our findings suggest that the formation of regular secondary structure is an intrinsic feature of random amino acid sequences, while the formation of long-disordered regions is not an intrinsic feature of proteins with disordered regions. Put differently, helices and strands appear to be maintained easily by evolution, whereas maintaining disordered regions appears difficult. Neutral mutations with respect to disorder are therefore very unlikely.
Figures
Similar articles
-
The unfoldomics decade: an update on intrinsically disordered proteins.BMC Genomics. 2008 Sep 16;9 Suppl 2(Suppl 2):S1. doi: 10.1186/1471-2164-9-S2-S1. BMC Genomics. 2008. PMID: 18831774 Free PMC article.
-
Sequence fingerprints distinguish erroneous from correct predictions of intrinsically disordered protein regions.J Biomol Struct Dyn. 2018 Dec;36(16):4338-4351. doi: 10.1080/07391102.2017.1415822. Epub 2017 Dec 27. J Biomol Struct Dyn. 2018. PMID: 29228892
-
Bioinformatical approaches to characterize intrinsically disordered/unstructured proteins.Brief Bioinform. 2010 Mar;11(2):225-43. doi: 10.1093/bib/bbp061. Epub 2009 Dec 10. Brief Bioinform. 2010. PMID: 20007729 Review.
-
The s2D method: simultaneous sequence-based prediction of the statistical populations of ordered and disordered regions in proteins.J Mol Biol. 2015 Feb 27;427(4):982-996. doi: 10.1016/j.jmb.2014.12.007. Epub 2014 Dec 20. J Mol Biol. 2015. PMID: 25534081
-
A practical overview of protein disorder prediction methods.Proteins. 2006 Oct 1;65(1):1-14. doi: 10.1002/prot.21075. Proteins. 2006. PMID: 16856179 Review.
Cited by
-
Evolution of Intrinsic Disorder in Protein Loops.Life (Basel). 2023 Oct 14;13(10):2055. doi: 10.3390/life13102055. Life (Basel). 2023. PMID: 37895436 Free PMC article.
-
Integration of new genes into cellular networks, and their structural maturation.Genetics. 2013 Dec;195(4):1407-17. doi: 10.1534/genetics.113.152256. Epub 2013 Sep 20. Genetics. 2013. PMID: 24056411 Free PMC article.
-
Proteome-wide evidence for enhanced positive Darwinian selection within intrinsically disordered regions in proteins.Genome Biol. 2011 Jul 19;12(7):R65. doi: 10.1186/gb-2011-12-7-r65. Genome Biol. 2011. PMID: 21771306 Free PMC article.
-
Large extent of disorder in Adenomatous Polyposis Coli offers a strategy to guard Wnt signalling against point mutations.PLoS One. 2013 Oct 9;8(10):e77257. doi: 10.1371/journal.pone.0077257. eCollection 2013. PLoS One. 2013. PMID: 24130866 Free PMC article.
-
The FCS-like zinc finger scaffold of the kinase SnRK1 is formed by the coordinated actions of the FLZ domain and intrinsically disordered regions.J Biol Chem. 2018 Aug 24;293(34):13134-13150. doi: 10.1074/jbc.RA118.002073. Epub 2018 Jun 26. J Biol Chem. 2018. PMID: 29945970 Free PMC article.
References
-
- Abagyan RA, Batalov S. Do aligned sequences share the same fold? J. Mol. Biol. 1997;273:355–368. - PubMed
-
- Anfinsen CB, Scheraga HA. Experimental and theoretical aspects of protein folding. Adv. Prot. Chem. 1975;29:205–300. - PubMed
-
- Benner SA, et al. Bona fide predictions of protein secondary structure using transparent analyses of multiple sequence alignments. Chem. Rev. 1997;97:2725–2844. - PubMed
