

Cellular signaling and NO production
- PMID: 20082095
- PMCID: PMC3774002
- DOI: 10.1007/s00424-009-0765-9
Cellular signaling and NO production
Abstract
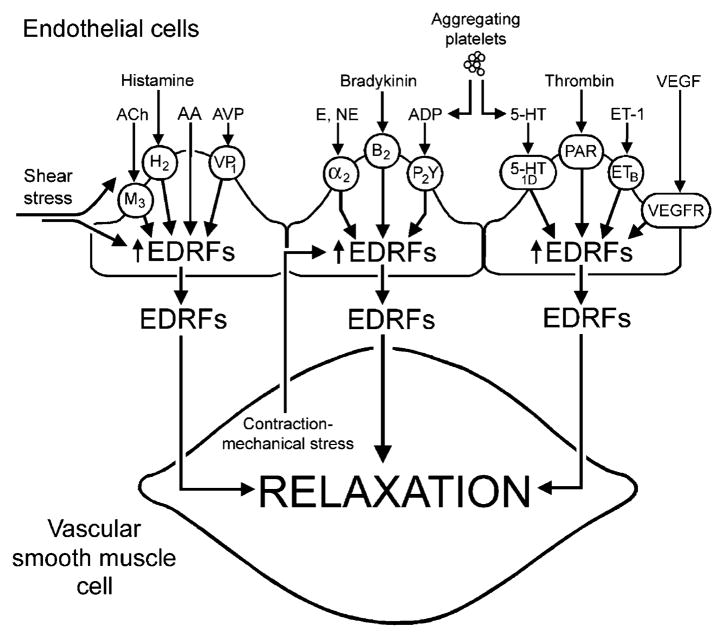
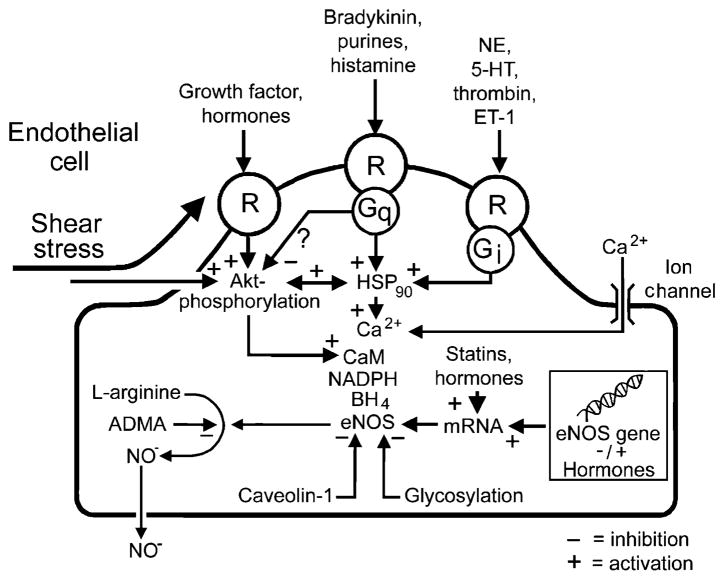
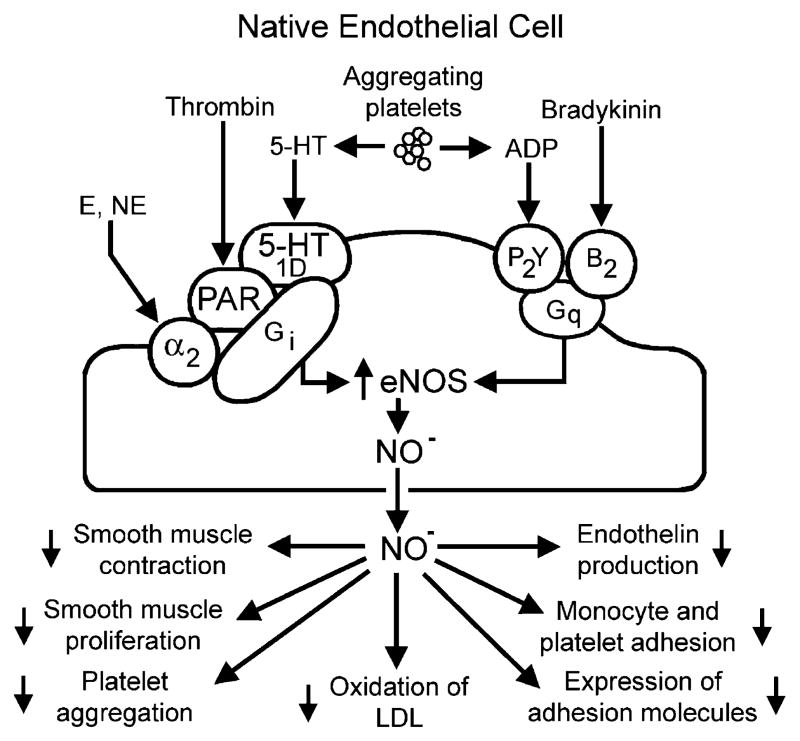
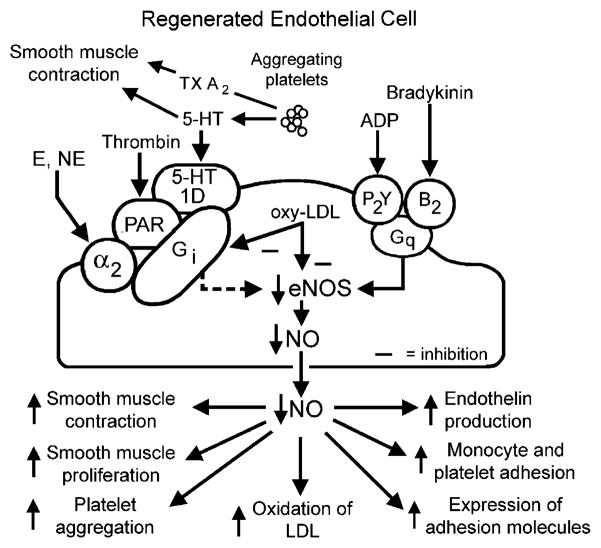
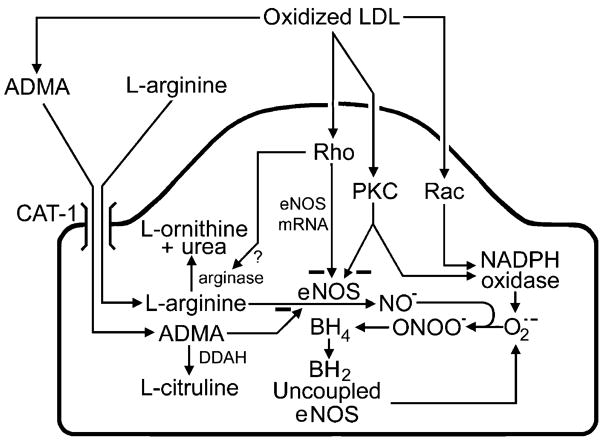
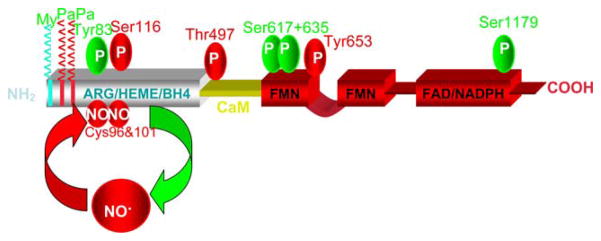
The endothelium can evoke relaxations (dilatations) of the underlying vascular smooth muscle, by releasing vasodilator substances. The best characterized endothelium-derived relaxing factor is nitric oxide (NO), which is synthesized by the endothelial isoform of nitric oxide synthase (eNOS). Endothelium-dependent relaxations involve both pertussis-toxin-sensitive G(i) (e.g., responses to serotonin, sphingosine 1-phosphate, alpha(2)-adrenergic agonists, and thrombin) and pertussis-toxin-insensitive G(q) (e.g., adenosine diphosphate and bradykinin) coupling proteins. eNOS undergoes a complex pattern of intracellular regulation, including post-translational modifications involving enzyme acylation and phosphorylation. eNOS is reversibly targeted to signal-transducing plasmalemmal caveolae where the enzyme interacts with a number of regulatory proteins, many of which are modified in cardiovascular disease states. The release of nitric oxide by the endothelial cell can be up- (e.g., by estrogens, exercise, and dietary factors) and down-regulated (e.g. oxidative stress, smoking, and oxidized low-density lipoproteins). It is reduced in the course of vascular disease (e.g., diabetes and hypertension). Arteries covered with regenerated endothelium (e.g. following angioplasty) selectively lose the pertussis-toxin-sensitive pathway for NO release which favors vasospasm, thrombosis, penetration of macrophages, cellular growth, and the inflammatory reaction leading to atherosclerosis. The unraveling of the complex interaction of the pathways regulating the presence and the activity of eNOS will enhance the understanding of the perturbations in endothelium-dependent signaling that are seen in cardiovascular disease states, and may lead to the identification of novel targets for therapeutic intervention.
Figures
Similar articles
-
Endothelial dysfunction and vascular disease - a 30th anniversary update.Acta Physiol (Oxf). 2017 Jan;219(1):22-96. doi: 10.1111/apha.12646. Epub 2016 Jan 25. Acta Physiol (Oxf). 2017. PMID: 26706498 Review.
-
Regeneration of the endothelium in vascular injury.Cardiovasc Drugs Ther. 2010 Aug;24(4):299-303. doi: 10.1007/s10557-010-6257-5. Cardiovasc Drugs Ther. 2010. PMID: 20689986 Review.
-
Endothelial dysfunction and vascular disease.Acta Physiol (Oxf). 2009 Jun;196(2):193-222. doi: 10.1111/j.1748-1716.2009.01964.x. Epub 2009 Feb 10. Acta Physiol (Oxf). 2009. PMID: 19220204 Review.
-
Endothelium-derived nitric oxide and vascular physiology and pathology.Cell Mol Life Sci. 1999 Jul;55(8-9):1078-87. doi: 10.1007/s000180050358. Cell Mol Life Sci. 1999. PMID: 10442089 Free PMC article. Review.
-
Targeting and translocation of endothelial nitric oxide synthase.Braz J Med Biol Res. 1999 Nov;32(11):1361-6. doi: 10.1590/s0100-879x1999001100006. Braz J Med Biol Res. 1999. PMID: 10559837 Review.
Cited by
-
γ-secretase inhibitor up-regulates vascular endothelial growth factor receptor-2 and endothelial nitric oxide synthase.Exp Ther Med. 2011 Jul;2(4):725-729. doi: 10.3892/etm.2011.257. Epub 2011 Apr 19. Exp Ther Med. 2011. PMID: 22977566 Free PMC article.
-
2-(2,4-dihydroxyphenyl)-5-(E)-propenylbenzofuran promotes endothelial nitric oxide synthase activity in human endothelial cells.Biochem Pharmacol. 2012 Sep 15;84(6):804-12. doi: 10.1016/j.bcp.2012.06.029. Epub 2012 Jul 6. Biochem Pharmacol. 2012. PMID: 22771373 Free PMC article.
-
Nitric Oxide Regulates Estrus Cycle Dependent Colonic Motility in Mice.Front Neurosci. 2021 Sep 29;15:647555. doi: 10.3389/fnins.2021.647555. eCollection 2021. Front Neurosci. 2021. PMID: 34658750 Free PMC article.
-
Wall stretch and thromboxane A₂ activate NO synthase (eNOS) in pulmonary arterial smooth muscle cells via H₂O₂ and Akt-dependent phosphorylation.Pflugers Arch. 2016 Apr;468(4):705-16. doi: 10.1007/s00424-015-1778-1. Epub 2016 Jan 4. Pflugers Arch. 2016. PMID: 26729266
-
The 24-h activity cycle and cardiovascular outcomes: establishing biological plausibility using arterial stiffness as an intermediate outcome.Am J Physiol Heart Circ Physiol. 2023 Dec 1;325(6):H1243-H1263. doi: 10.1152/ajpheart.00258.2023. Epub 2023 Sep 22. Am J Physiol Heart Circ Physiol. 2023. PMID: 37737729 Free PMC article. Review.
References
-
- Aikawa M, Libby P. The vulnerable atherosclerotic plaque pathogenesis and therapeutic approach. Cardiovasc Path. 2004;13:125–138. - PubMed
-
- Balligand JL, Feron O, et al. eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev. 2009;89(2):481–534. - PubMed
-
- Berka V, Wu G, Yeh HC, et al. Three different oxygen-induced radical species in endothelial nitric-oxide synthase oxygenase domain under regulation by L-arginine and tetrahydrobiopterin. J Biol Chem. 2004;279:32243–32251. - PubMed
-
- Busse R, Edwards G, Félétou M, Fleming I, Vanhoutte PM. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–380. - PubMed
-
- Busse R, Fleming I. Regulation of endothelium-derived vasoactive autacoid production by hemodynamic forces. Trends Pharmacol Sci. 2003;24:24–29. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
