

A CD1d-dependent antagonist inhibits the activation of invariant NKT cells and prevents development of allergen-induced airway hyperreactivity
- PMID: 20083656
- PMCID: PMC2845715
- DOI: 10.4049/jimmunol.0901208
A CD1d-dependent antagonist inhibits the activation of invariant NKT cells and prevents development of allergen-induced airway hyperreactivity
Abstract
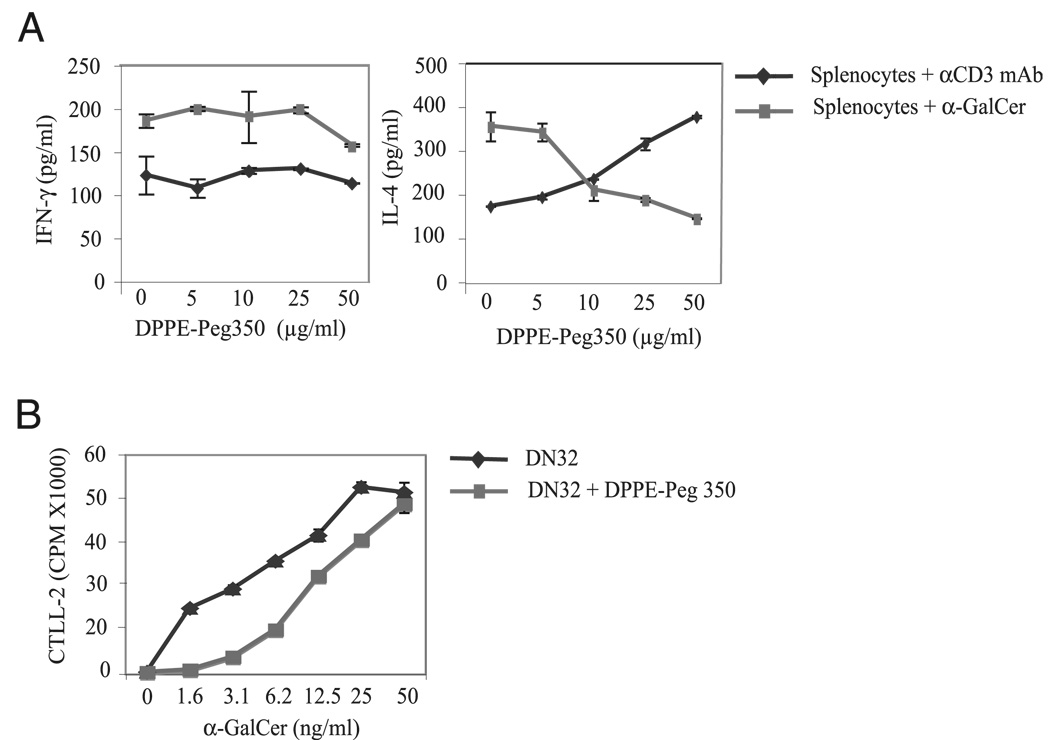
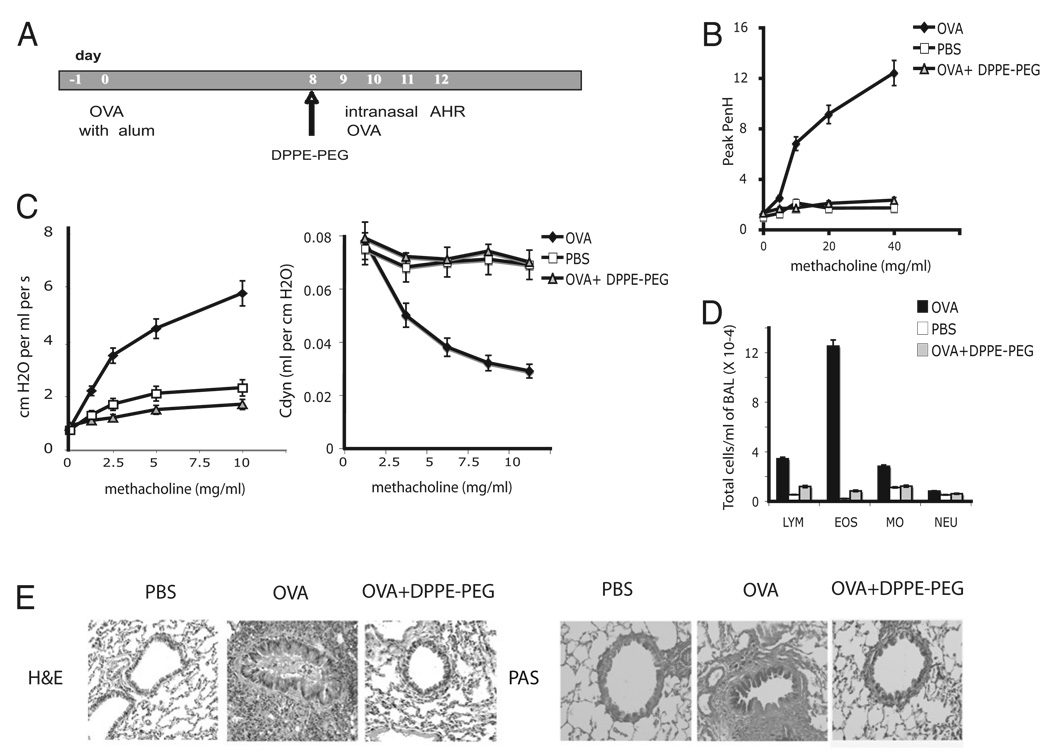
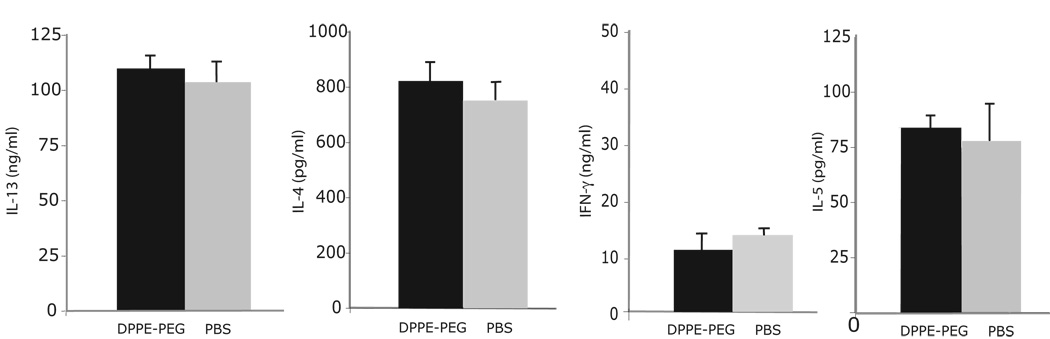
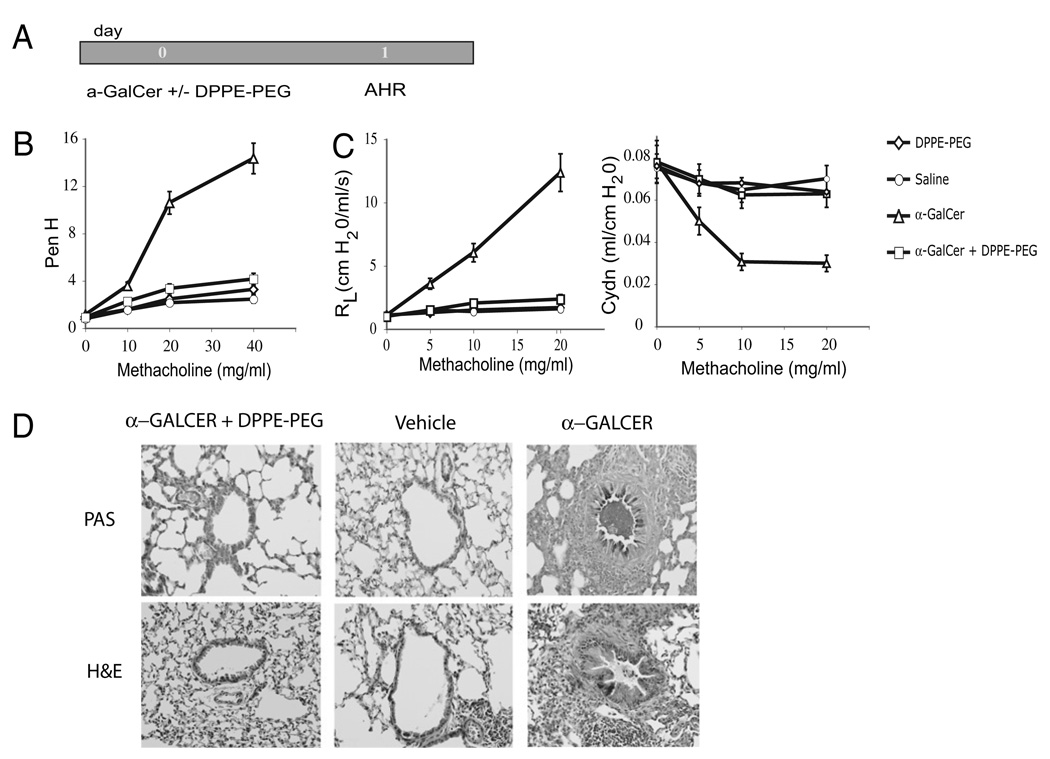
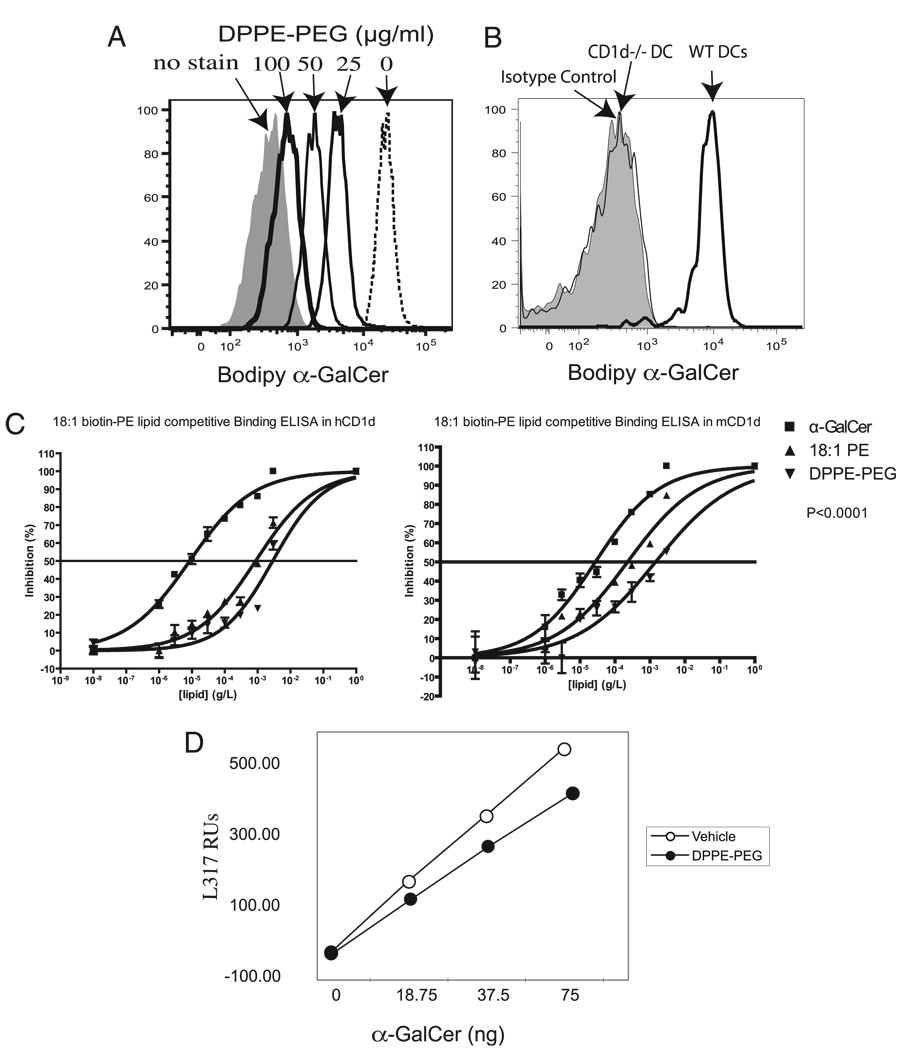
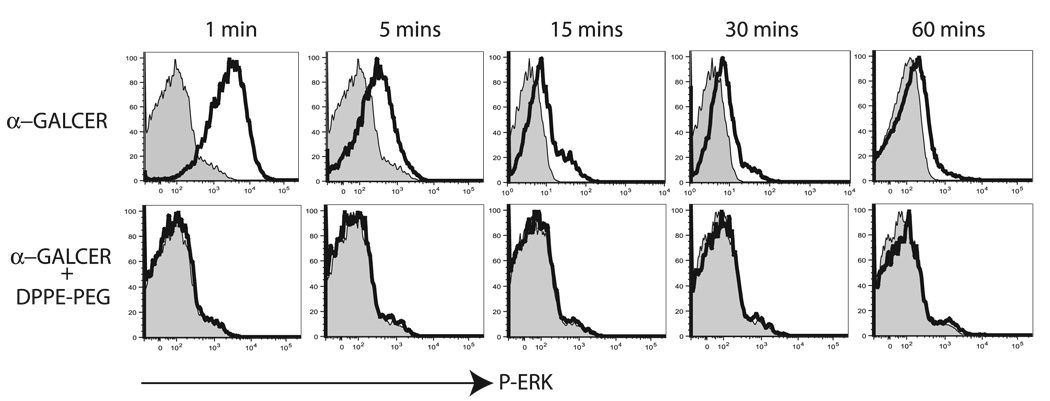
The prevalence of asthma continues to increase in westernized countries, and optimal treatment remains a significant therapeutic challenge. Recently, CD1d-restricted invariant NKT (iNKT) cells were found to play a critical role in the induction of airway hyperreactivity (AHR) in animal models and are associated with asthma in humans. To test whether iNKT cell-targeted therapy could be used to treat allergen-induced airway disease, mice were sensitized with OVA and treated with di-palmitoyl-phosphatidyl-ethanolamine polyethylene glycol (DPPE-PEG), a CD1d-binding lipid antagonist. A single dose of DPPE-PEG prevented the development of AHR and pulmonary infiltration of lymphocytes upon OVA challenge, but had no effect on the development of OVA-specific Th2 responses. In addition, DPPE-PEG completely prevented the development of AHR after administration of alpha-galactosylceramide (alpha-GalCer) intranasally. Furthermore, we demonstrate that DPPE-PEG acts as antagonist to alpha-GalCer and competes with alpha-GalCer for binding to CD1d. Finally, we show that DPPE-PEG completely inhibits the alpha-GalCer-induced phosphorylation of ERK tyrosine kinase in iNKT cells, suggesting that DPPE-PEG specifically blocks TCR signaling and thus activation of iNKT cells. Because iNKT cells play a critical role in the development of AHR, the inhibition of iNKT activation by DPPE-PEG suggests a novel approach to treat iNKT cell-mediated diseases such as asthma.
Figures
References
-
- Centers for Disease Control and Prevention (CDC) Forecasted state-specific estimates of self-reported asthma prevalence—United States, 1998. MMWR Morb. Mortal. Wkly. Rep. 1998;47:1022–1025. - PubMed
-
- Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu. Rev. Immunol. 1999;17:255–281. - PubMed
-
- Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N. Engl. J. Med. 1992;326:298–304. - PubMed
-
- Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
