

Multi-platform whole-genome microarray analyses refine the epigenetic signature of breast cancer metastasis with gene expression and copy number
- PMID: 20084286
- PMCID: PMC2801616
- DOI: 10.1371/journal.pone.0008665
Multi-platform whole-genome microarray analyses refine the epigenetic signature of breast cancer metastasis with gene expression and copy number
Abstract
Background: We have previously identified genome-wide DNA methylation changes in a cell line model of breast cancer metastasis. These complex epigenetic changes that we observed, along with concurrent karyotype analyses, have led us to hypothesize that complex genomic alterations in cancer cells (deletions, translocations and ploidy) are superimposed over promoter-specific methylation events that are responsible for gene-specific expression changes observed in breast cancer metastasis.
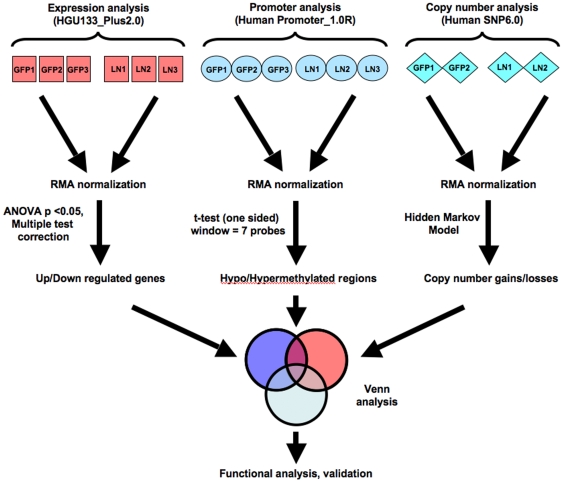
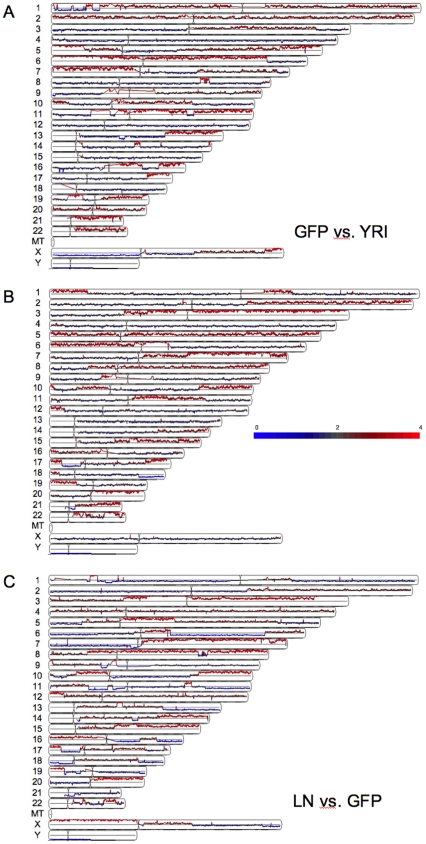
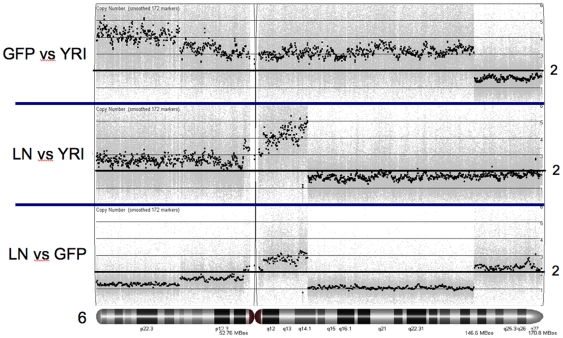
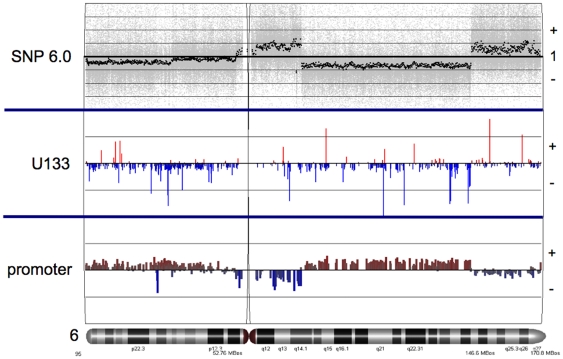
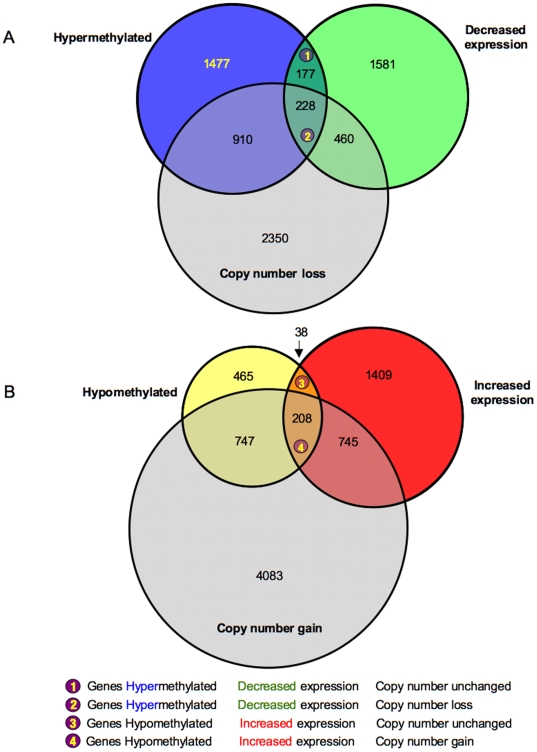
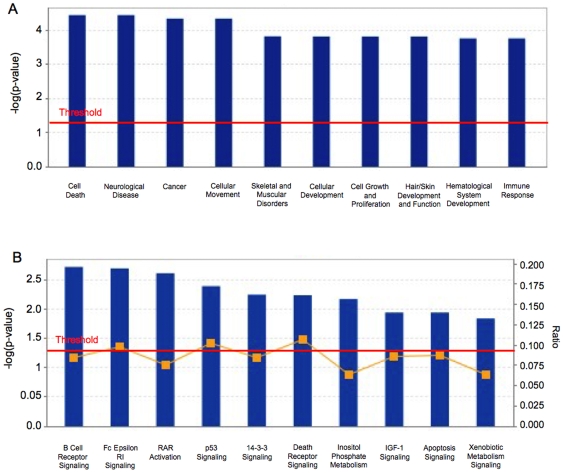
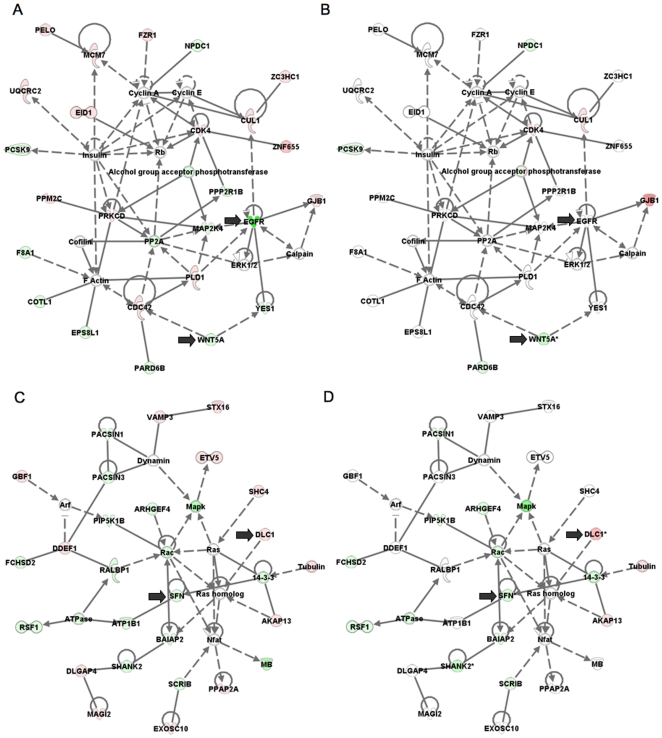
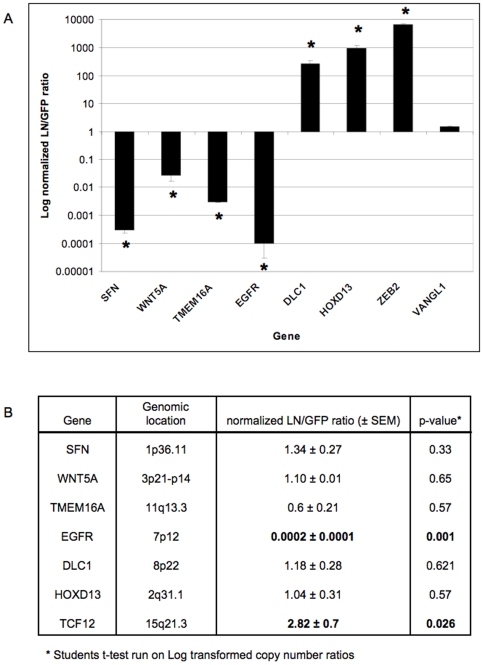
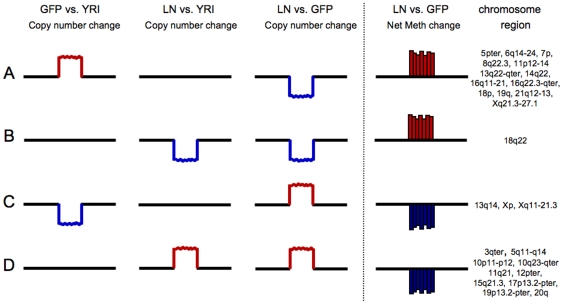
Methodology/principal findings: We undertook simultaneous high-resolution, whole-genome analyses of MDA-MB-468GFP and MDA-MB-468GFP-LN human breast cancer cell lines (an isogenic, paired lymphatic metastasis cell line model) using Affymetrix gene expression (U133), promoter (1.0R), and SNP/CNV (SNP 6.0) microarray platforms to correlate data from gene expression, epigenetic (DNA methylation), and combination copy number variant/single nucleotide polymorphism microarrays. Using Partek Software and Ingenuity Pathway Analysis we integrated datasets from these three platforms and detected multiple hypomethylation and hypermethylation events. Many of these epigenetic alterations correlated with gene expression changes. In addition, gene dosage events correlated with the karyotypic differences observed between the cell lines and were reflected in specific promoter methylation patterns. Gene subsets were identified that correlated hyper (and hypo) methylation with the loss (or gain) of gene expression and in parallel, with gene dosage losses and gains, respectively. Individual gene targets from these subsets were also validated for their methylation, expression and copy number status, and susceptible gene pathways were identified that may indicate how selective advantage drives the processes of tumourigenesis and metastasis.
Conclusions/significance: Our approach allows more precisely profiling of functionally relevant epigenetic signatures that are associated with cancer progression and metastasis.
Conflict of interest statement
Figures


References
-
- Xu J, Chambers AF, Tuck AB, Rodenhiser DI. Molecular cytogenetic characterization of human breast cancer cell line MDA-MB-468 and its variant 468LN, which displays aggressive lymphatic metastasis. Cancer Genetics and Cytogenetics. 2008;181:1–7. - PubMed
-
- van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. - PubMed
-
- Sadikovic B, Andrews J, Carter D, Robinson J, Rodenhiser DI. Genome-wide H3K9 histone acetylation profiles are altered in benzopyrene treated MCF7 breast cancer cells. J Biol Chem. 2008;283:4051–4060. - PubMed
-
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
