

Rapid genomic characterization of the genus vitis
- PMID: 20084295
- PMCID: PMC2805708
- DOI: 10.1371/journal.pone.0008219
Rapid genomic characterization of the genus vitis
Abstract
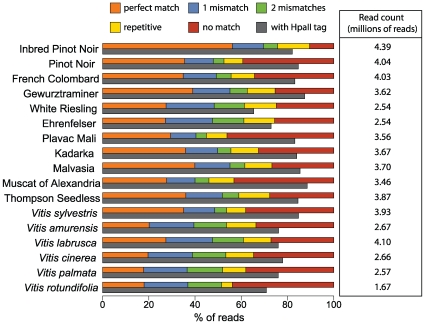
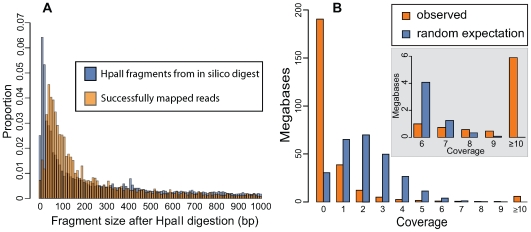
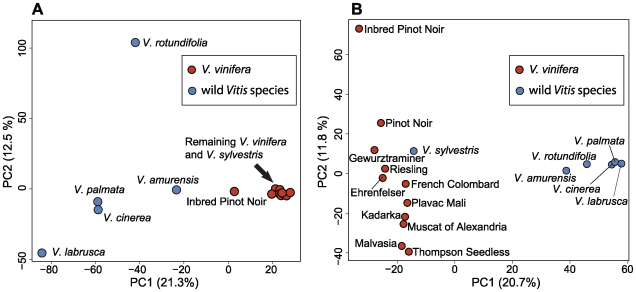
Next-generation sequencing technologies promise to dramatically accelerate the use of genetic information for crop improvement by facilitating the genetic mapping of agriculturally important phenotypes. The first step in optimizing the design of genetic mapping studies involves large-scale polymorphism discovery and a subsequent genome-wide assessment of the population structure and pattern of linkage disequilibrium (LD) in the species of interest. In the present study, we provide such an assessment for the grapevine (genus Vitis), the world's most economically important fruit crop. Reduced representation libraries (RRLs) from 17 grape DNA samples (10 cultivated V. vinifera and 7 wild Vitis species) were sequenced with sequencing-by-synthesis technology. We developed heuristic approaches for SNP calling, identified hundreds of thousands of SNPs and validated a subset of these SNPs on a 9K genotyping array. We demonstrate that the 9K SNP array provides sufficient resolution to distinguish among V. vinifera cultivars, between V. vinifera and wild Vitis species, and even among diverse wild Vitis species. We show that there is substantial sharing of polymorphism between V. vinifera and wild Vitis species and find that genetic relationships among V. vinifera cultivars agree well with their proposed geographic origins using principal components analysis (PCA). Levels of LD in the domesticated grapevine are low even at short ranges, but LD persists above background levels to 3 kb. While genotyping arrays are useful for assessing population structure and the decay of LD across large numbers of samples, we suggest that whole-genome sequencing will become the genotyping method of choice for genome-wide genetic mapping studies in high-diversity plant species. This study demonstrates that we can move quickly towards genome-wide studies of crop species using next-generation sequencing. Our study sets the stage for future work in other high diversity crop species, and provides a significant enhancement to current genetic resources available to the grapevine genetic community.
Conflict of interest statement
Figures



Similar articles
-
High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology.BMC Genomics. 2007 Nov 19;8:424. doi: 10.1186/1471-2164-8-424. BMC Genomics. 2007. PMID: 18021442 Free PMC article.
-
SNP-Discovery by RAD-Sequencing in a Germplasm Collection of Wild and Cultivated Grapevines (V. vinifera L.).PLoS One. 2017 Jan 26;12(1):e0170655. doi: 10.1371/journal.pone.0170655. eCollection 2017. PLoS One. 2017. PMID: 28125640 Free PMC article.
-
Extended diversity analysis of cultivated grapevine Vitis vinifera with 10K genome-wide SNPs.PLoS One. 2018 Feb 8;13(2):e0192540. doi: 10.1371/journal.pone.0192540. eCollection 2018. PLoS One. 2018. PMID: 29420602 Free PMC article.
-
Review: Status and prospects of association mapping in grapevine.Plant Sci. 2023 Feb;327:111539. doi: 10.1016/j.plantsci.2022.111539. Epub 2022 Nov 21. Plant Sci. 2023. PMID: 36410567 Review.
-
Vitis vinifera genotyping toolbox to highlight diversity and germplasm identification.Front Plant Sci. 2023 Apr 26;14:1139647. doi: 10.3389/fpls.2023.1139647. eCollection 2023. Front Plant Sci. 2023. PMID: 37180393 Free PMC article. Review.
Cited by
-
NGS technologies for analyzing germplasm diversity in genebanks.Brief Funct Genomics. 2012 Jan;11(1):38-50. doi: 10.1093/bfgp/elr046. Epub 2012 Jan 17. Brief Funct Genomics. 2012. PMID: 22257472 Free PMC article. Review.
-
A candidate gene association study on muscat flavor in grapevine (Vitis vinifera L.).BMC Plant Biol. 2010 Nov 9;10:241. doi: 10.1186/1471-2229-10-241. BMC Plant Biol. 2010. PMID: 21062440 Free PMC article.
-
Facing Climate Change: Biotechnology of Iconic Mediterranean Woody Crops.Front Plant Sci. 2019 Apr 16;10:427. doi: 10.3389/fpls.2019.00427. eCollection 2019. Front Plant Sci. 2019. PMID: 31057569 Free PMC article. Review.
-
Anti-inflammatory effects of Vitis thunbergii var. taiwaniana on knee damage associated with arthritis.J Med Food. 2014 Apr;17(4):479-86. doi: 10.1089/jmf.2013.2914. J Med Food. 2014. PMID: 24720858 Free PMC article.
-
Development and validation of a 20K single nucleotide polymorphism (SNP) whole genome genotyping array for apple (Malus × domestica Borkh).PLoS One. 2014 Oct 10;9(10):e110377. doi: 10.1371/journal.pone.0110377. eCollection 2014. PLoS One. 2014. PMID: 25303088 Free PMC article.
References
-
- Mackay TF, Stone EA, Ayroles JF. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 2009;10:565–577. - PubMed
-
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356. - PubMed
-
- Heffner EL, Sorrells ME, Jannink J-L. Genomic Selection for Crop Improvement. Crop Sci. 2009;49:1–12.
-
- Nordborg M, Weigel D. Next-generation genetics in plants. Nature. 2008;456:720–723. - PubMed
-
- Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, et al. Whole-genome sequencing and variant discovery in C. elegans. Nat Meth. 2008;5:183. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
