

Comparative metagenomic analysis of plasmid encoded functions in the human gut microbiome
- PMID: 20085629
- PMCID: PMC2822762
- DOI: 10.1186/1471-2164-11-46
Comparative metagenomic analysis of plasmid encoded functions in the human gut microbiome
Abstract
Background: Little is known regarding the pool of mobile genetic elements associated with the human gut microbiome. In this study we employed the culture independent TRACA system to isolate novel plasmids from the human gut microbiota, and a comparative metagenomic analysis to investigate the distribution and relative abundance of functions encoded by these plasmids in the human gut microbiome.
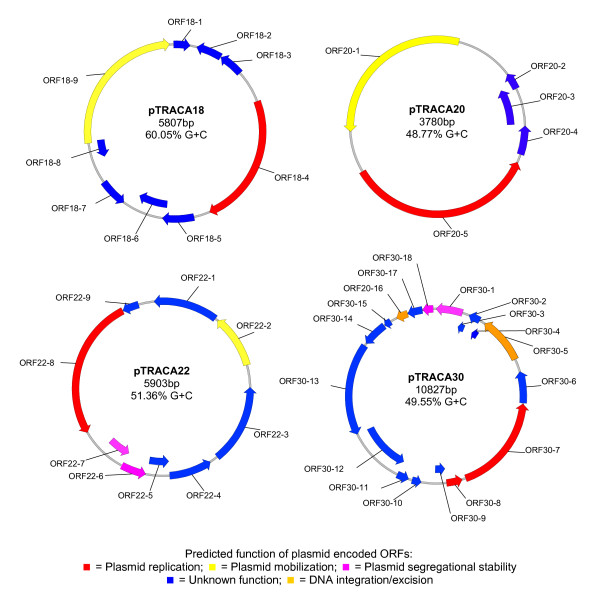
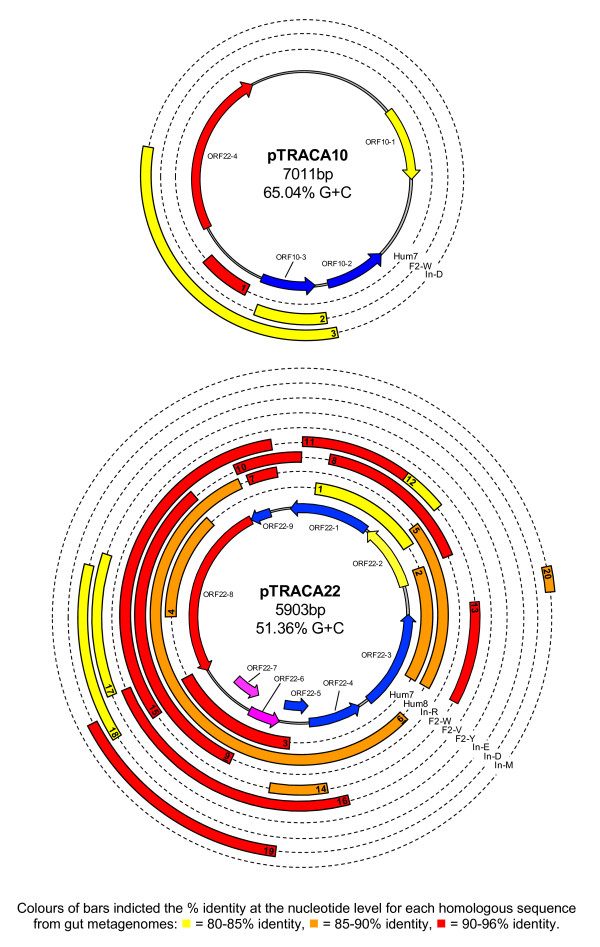
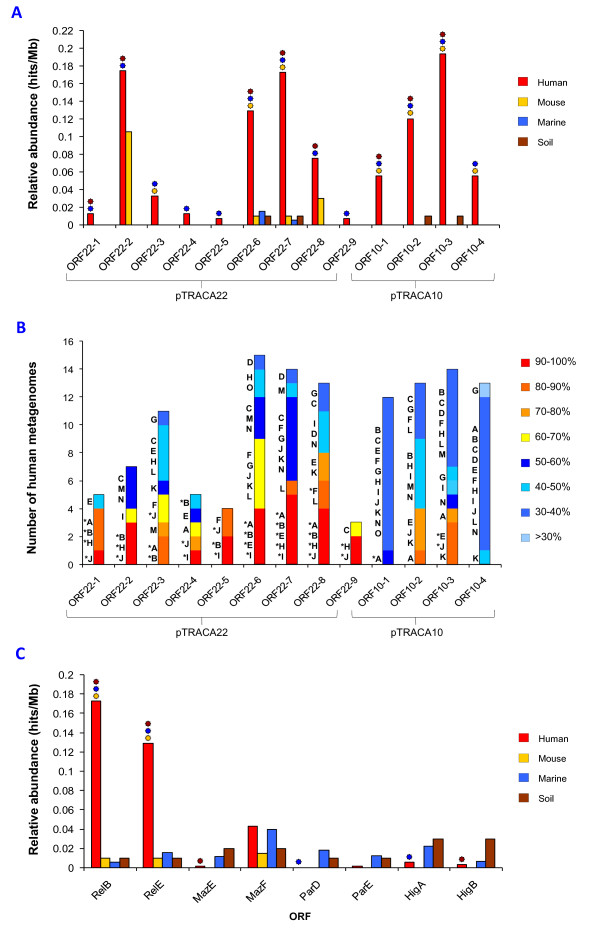
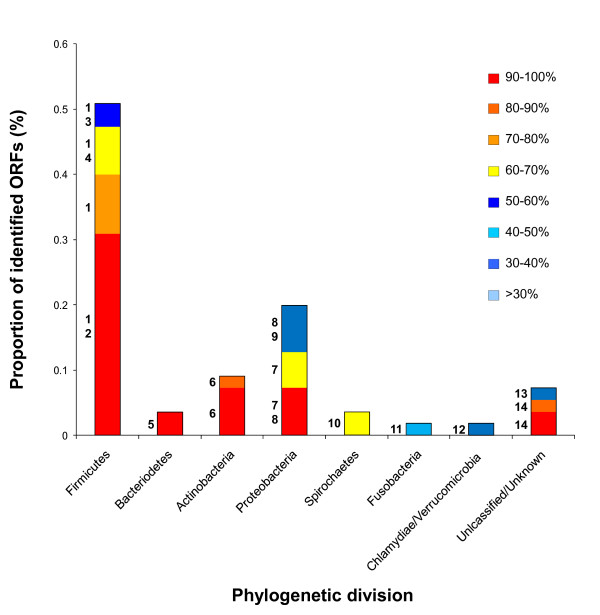
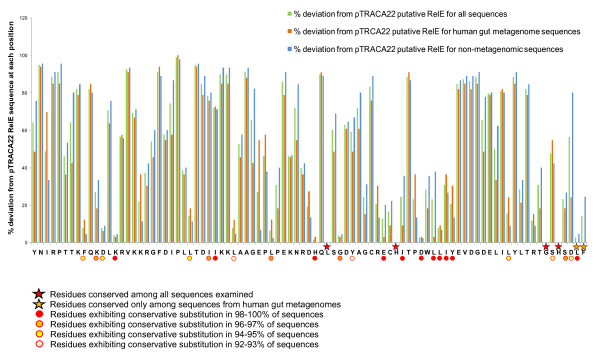
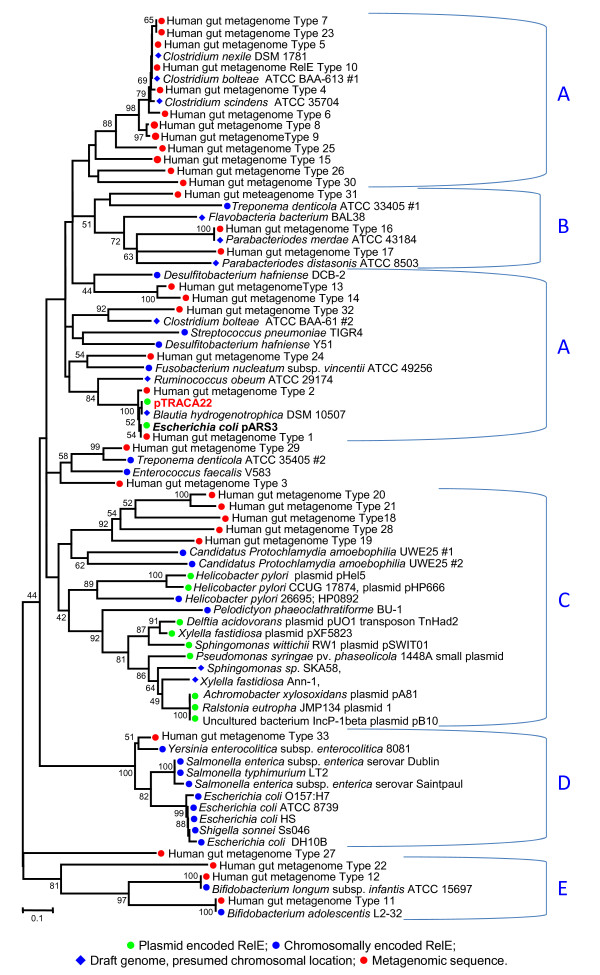
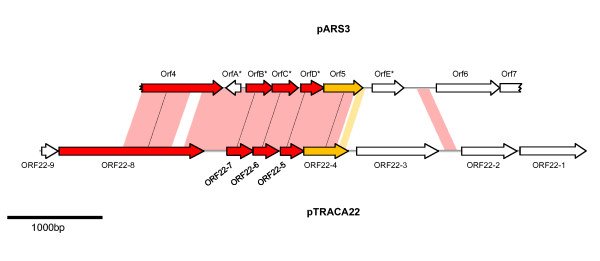
Results: Novel plasmids were acquired from the human gut microbiome, and homologous nucleotide sequences with high identity (>90%) to two plasmids (pTRACA10 and pTRACA22) were identified in the multiple human gut microbiomes analysed here. However, no homologous nucleotide sequences to these plasmids were identified in the murine gut or environmental metagenomes. Functions encoded by the plasmids pTRACA10 and pTRACA22 were found to be more prevalent in the human gut microbiome when compared to microbial communities from other environments. Among the most prevalent functions identified was a putative RelBE toxin-antitoxin (TA) addiction module, and subsequent analysis revealed that this was most closely related to putative TA modules from gut associated bacteria belonging to the Firmicutes. A broad phylogenetic distribution of RelE toxin genes was observed in gut associated bacterial species (Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria), but no RelE homologues were identified in gut associated archaeal species. We also provide indirect evidence for the horizontal transfer of these genes between bacterial species belonging to disparate phylogenetic divisions, namely Gram negative Proteobacteria and Gram positive species from the Firmicutes division.
Conclusions: The application of a culture independent system to capture novel plasmids from the human gut mobile metagenome, coupled with subsequent comparative metagenomic analysis, highlighted the unexpected prevalence of plasmid encoded functions in the gut microbial ecosystem. In particular the increased relative abundance and broad phylogenetic distribution was identified for a putative RelBE toxin/antitoxin addiction module, a putative phosphohydrolase/phosphoesterase, and an ORF of unknown function. Our analysis also indicates that some plasmids or plasmid families are present in the gut microbiomes of geographically isolated human hosts with a broad global distribution (America, Japan and Europe), and are potentially unique to the human gut microbiome. Further investigation of the plasmid population associated with the human gut is likely to provide important insights into the development, functioning and evolution of the human gut microbiota.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
