

Leveraging genetic variability across populations for the identification of causal variants
- PMID: 20085711
- PMCID: PMC2801753
- DOI: 10.1016/j.ajhg.2009.11.016
Leveraging genetic variability across populations for the identification of causal variants
Abstract
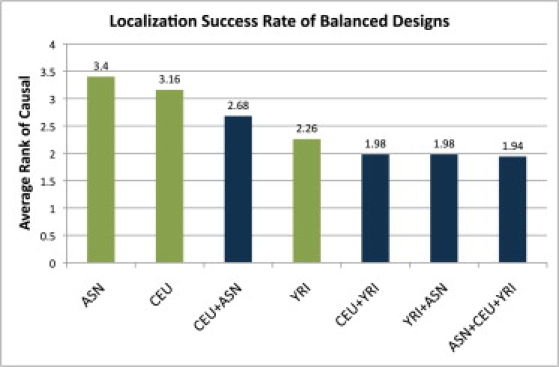
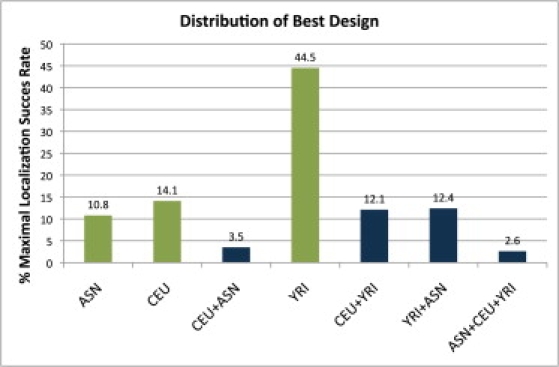
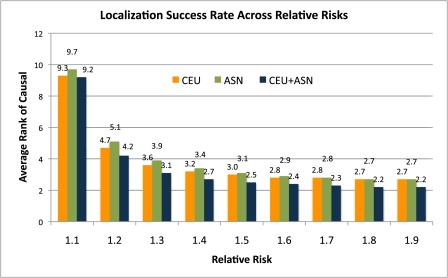
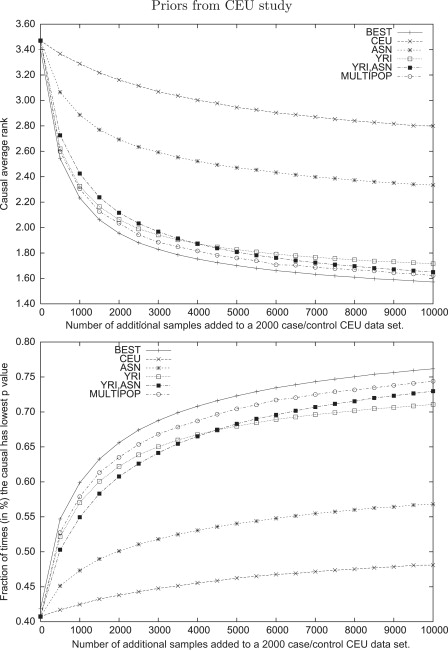
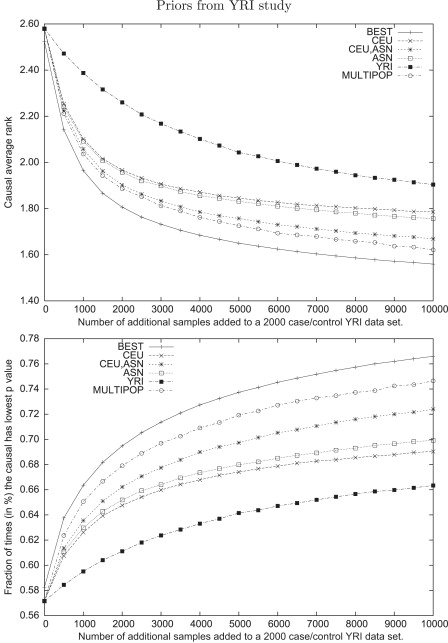
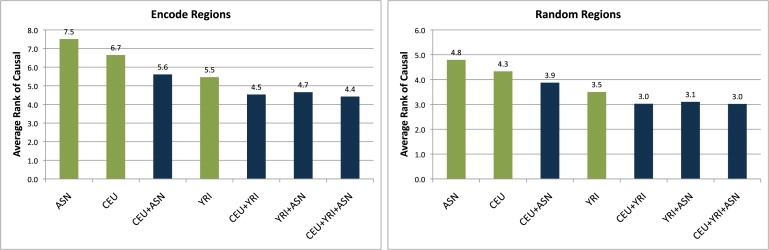
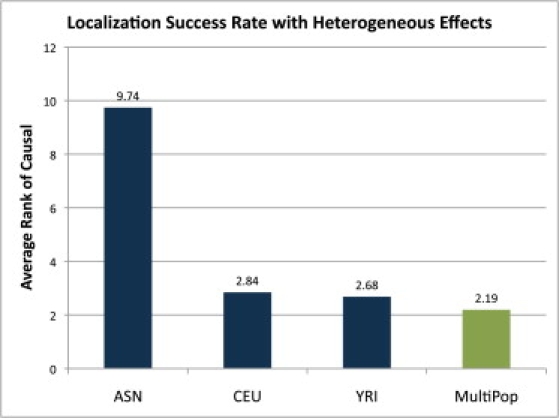
Genome-wide association studies have been performed extensively in the last few years, resulting in many new discoveries of genomic regions that are associated with complex traits. It is often the case that a SNP found to be associated with the condition is not the causal SNP, but a proxy to it as a result of linkage disequilibrium. For the identification of the actual causal SNP, fine-mapping follow-up is performed, either with the use of dense genotyping or by sequencing of the region. In either case, if the causal SNP is in high linkage disequilibrium with other SNPs, the fine-mapping procedure will require a very large sample size for the identification of the causal SNP. Here, we show that by leveraging genetic variability across populations, we significantly increase the localization success rate (LSR) for a causal SNP in a follow-up study that involves multiple populations as compared to a study that involves only one population. Thus, the average power for detection of the causal variant will be higher in a joint analysis than that in studies in which only one population is analyzed at a time. On the basis of this observation, we developed a framework to efficiently search for a follow-up study design: our framework searches for the best combination of populations from a pool of available populations to maximize the LSR for detection of a causal variant. This framework and its accompanying software can be used to considerably enhance the power of fine-mapping studies.
2010 The American Society of Human Genetics. Published by Elsevier Inc.
Figures
References
-
- Udler M.S., Meyer K.B., Pooley K.A., Karlins E., Struewing J.P., Zhang J., Doody D.R., MacArthur S., Tyrer J., Pharoah P.D., SEARCH Collaborators FGFR2 variants and breast cancer risk: fine-scale mapping using African American studies and analysis of chromatin conformation. Hum. Mol. Genet. 2009;18:1692–1703. - PMC - PubMed
-
- Easton D.F., Pooley K.A., Dunning A.M., Pharoah P.D.P., Thompson D., Ballinger D.G., Struewing J.P., Morrison J., Field H., Luben R., SEARCH collaborators. kConFab. AOCS Management Group Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. - PMC - PubMed
-
- Ioannidis J.P.A., Ntzani E.E., Trikalinos T.A. ‘Racial’ differences in genetic effects for complex diseases. Nat. Genet. 2004;36:1312–1318. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
