

Role of calcium-independent phospholipase A2beta in high glucose-induced activation of RhoA, Rho kinase, and CPI-17 in cultured vascular smooth muscle cells and vascular smooth muscle hypercontractility in diabetic animals
- PMID: 20086008
- PMCID: PMC2838285
- DOI: 10.1074/jbc.M109.057711
Role of calcium-independent phospholipase A2beta in high glucose-induced activation of RhoA, Rho kinase, and CPI-17 in cultured vascular smooth muscle cells and vascular smooth muscle hypercontractility in diabetic animals
Abstract
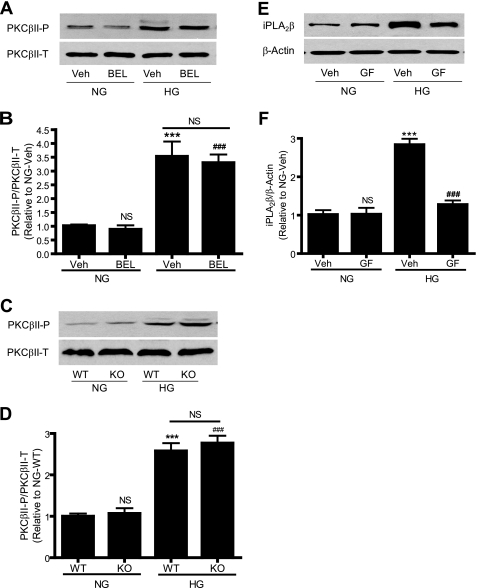
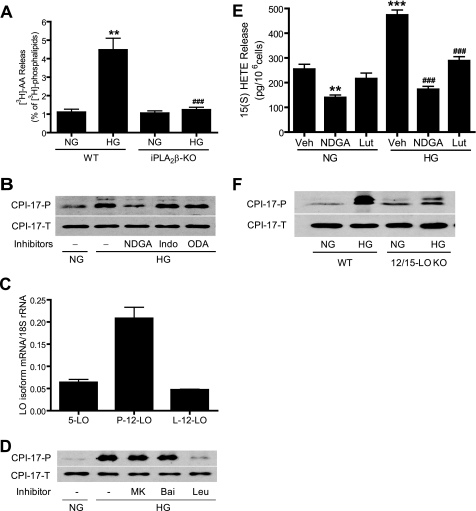
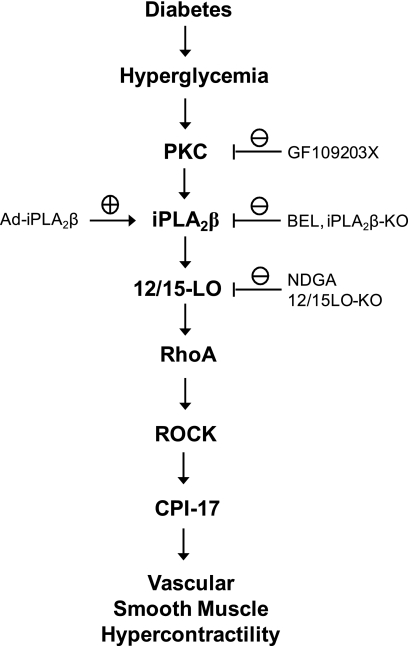
Previous studies suggest that high glucose-induced RhoA/Rho kinase/CPI-17 activation is involved in diabetes-associated vascular smooth muscle hypercontractility. However, the upstream signaling that links high glucose and RhoA/Rho kinase/CPI-17 activation is unknown. Here we report that calcium-independent phospholipase A(2)beta (iPLA(2)beta) is required for high glucose-induced RhoA/Rho kinase/CPI-17 activation and thereby contributes to diabetes-associated vascular smooth muscle hypercontractility. We demonstrate that high glucose increases iPLA(2)beta mRNA, protein, and iPLA(2) activity in a time-dependent manner. Protein kinase C is involved in high glucose-induced iPLA(2)beta protein up-regulation. Inhibiting iPLA(2)beta activity with bromoenol lactone or preventing its expression by genetic deletion abolishes high glucose-induced RhoA/Rho kinase/CPI-17 activation, and restoring expression of iPLA(2)beta in iPLA(2)beta-deficient cells also restores high glucose-induced CPI-17 phosphorylation. Pharmacological and genetic inhibition of 12/15-lipoxygenases has effects on high glucose-induced CPI-17 phosphorylation similar to iPLA(2)beta inhibition. Moreover, increases in iPLA(2) activity and iPLA(2)beta protein expression are also observed in both type 1 and type 2 diabetic vasculature. Pharmacological and genetic inhibition of iPLA(2)beta, but not iPLA(2)gamma, diminishes diabetes-associated vascular smooth muscle hypercontractility. In summary, our results reveal a novel mechanism by which high glucose-induced, protein kinase C-mediated iPLA(2)beta up-regulation activates the RhoA/Rho kinase/CPI-17 via 12/15-lipoxygenases and thereby contributes to diabetes-associated vascular smooth muscle hypercontractility.
Figures





Similar articles
-
Up-regulation of CPI-17 phosphorylation in diabetic vasculature and high glucose cultured vascular smooth muscle cells.Cardiovasc Res. 2006 Feb 1;69(2):491-501. doi: 10.1016/j.cardiores.2005.11.002. Epub 2005 Dec 5. Cardiovasc Res. 2006. PMID: 16336954
-
Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle.Circ Res. 2003 Mar 7;92(4):411-8. doi: 10.1161/01.RES.0000059987.90200.44. Epub 2003 Feb 6. Circ Res. 2003. PMID: 12600888
-
Increased basal phosphorylation of detrusor smooth muscle myosin in alloxan-induced diabetic rabbit is mediated by upregulation of Rho-kinase beta and CPI-17.Am J Physiol Renal Physiol. 2006 Mar;290(3):F650-6. doi: 10.1152/ajprenal.00235.2005. Epub 2005 Oct 4. Am J Physiol Renal Physiol. 2006. PMID: 16204412
-
Mechanisms underlying the pathogenesis of hyper-contractility of bronchial smooth muscle in allergic asthma.J Smooth Muscle Res. 2017;53(0):37-47. doi: 10.1540/jsmr.53.37. J Smooth Muscle Res. 2017. PMID: 28484126 Free PMC article. Review.
-
The role of RhoA and Rho-associated kinase in vascular smooth muscle contraction.Curr Hypertens Rep. 2003 Feb;5(1):66-72. doi: 10.1007/s11906-003-0013-1. Curr Hypertens Rep. 2003. PMID: 12530938 Review.
Cited by
-
Identification of a cAMP-response element in the regulator of G-protein signaling-2 (RGS2) promoter as a key cis-regulatory element for RGS2 transcriptional regulation by angiotensin II in cultured vascular smooth muscles.J Biol Chem. 2011 Dec 30;286(52):44646-58. doi: 10.1074/jbc.M111.265462. Epub 2011 Nov 4. J Biol Chem. 2011. PMID: 22057271 Free PMC article.
-
Effects of dietary palmitoleic acid on vascular function in aorta of diabetic mice.BMC Endocr Disord. 2022 Apr 18;22(1):103. doi: 10.1186/s12902-022-01018-2. BMC Endocr Disord. 2022. PMID: 35436932 Free PMC article.
-
Group VIA PLA2 (iPLA2β) is activated upstream of p38 mitogen-activated protein kinase (MAPK) in pancreatic islet β-cell signaling.J Biol Chem. 2012 Feb 17;287(8):5528-41. doi: 10.1074/jbc.M111.285114. Epub 2011 Dec 22. J Biol Chem. 2012. PMID: 22194610 Free PMC article.
-
Alterations in β-Cell Sphingolipid Profile Associated with ER Stress and iPLA2β: Another Contributor to β-Cell Apoptosis in Type 1 Diabetes.Molecules. 2021 Oct 21;26(21):6361. doi: 10.3390/molecules26216361. Molecules. 2021. PMID: 34770770 Free PMC article.
-
Platelet-activating factor and metastasis: calcium-independent phospholipase A2β deficiency protects against breast cancer metastasis to the lung.Am J Physiol Cell Physiol. 2011 Apr;300(4):C825-32. doi: 10.1152/ajpcell.00502.2010. Epub 2011 Jan 12. Am J Physiol Cell Physiol. 2011. PMID: 21228317 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
