

Population differentiation as a test for selective sweeps
- PMID: 20086244
- PMCID: PMC2840981
- DOI: 10.1101/gr.100545.109
Population differentiation as a test for selective sweeps
Abstract
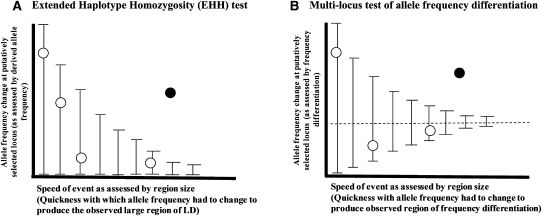
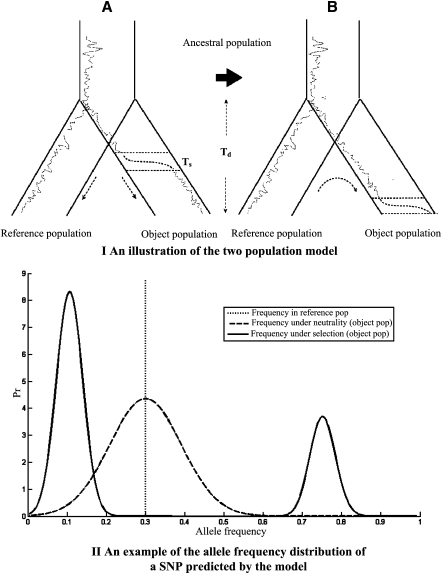
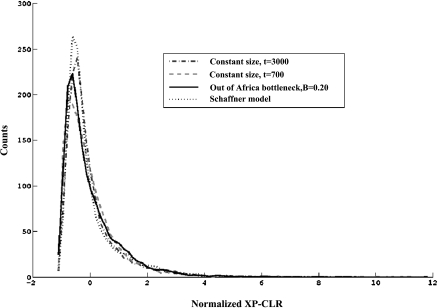
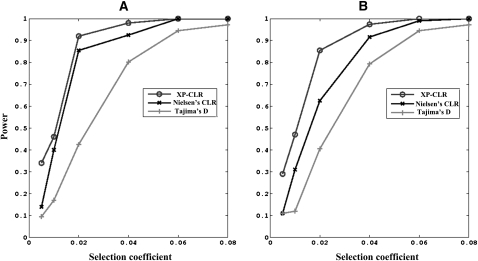
Selective sweeps can increase genetic differentiation among populations and cause allele frequency spectra to depart from the expectation under neutrality. We present a likelihood method for detecting selective sweeps that involves jointly modeling the multilocus allele frequency differentiation between two populations. We use Brownian motion to model genetic drift under neutrality, and a deterministic model to approximate the effect of a selective sweep on single nucleotide polymorphisms (SNPs) in the vicinity. We test the method with extensive simulated data, and demonstrate that in some scenarios the method provides higher power than previously reported approaches to detect selective sweeps, and can provide surprisingly good localization of the position of a selected allele. A strength of our technique is that it uses allele frequency differentiation between populations, which is much more robust to ascertainment bias in SNP discovery than methods based on the allele frequency spectrum. We apply this method to compare continentally diverse populations, as well as Northern and Southern Europeans. Our analysis identifies a list of loci as candidate targets of selection, including well-known selected loci and new regions that have not been highlighted by previous scans for selection.
Figures
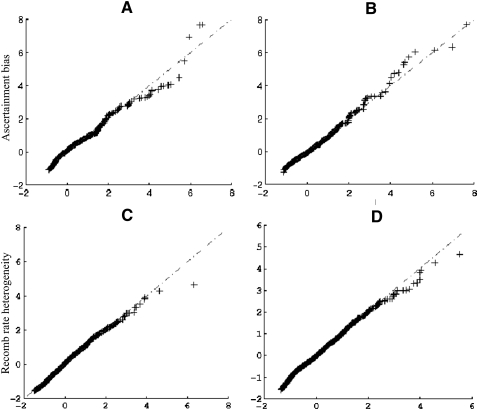
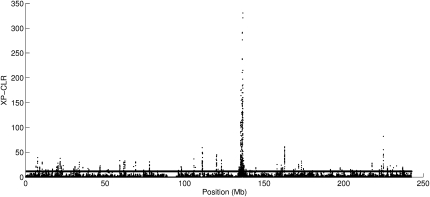
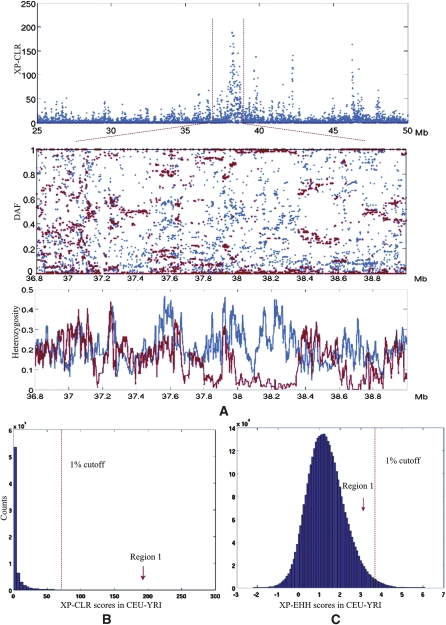
References
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources