

Role of Nogo-A in neuronal survival in the reperfused ischemic brain
- PMID: 20087369
- PMCID: PMC2949191
- DOI: 10.1038/jcbfm.2009.268
Role of Nogo-A in neuronal survival in the reperfused ischemic brain
Abstract
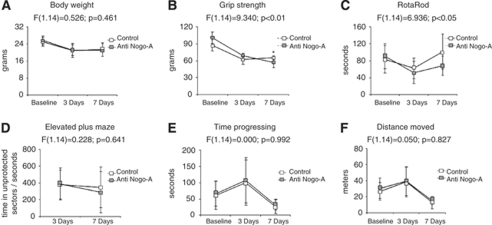
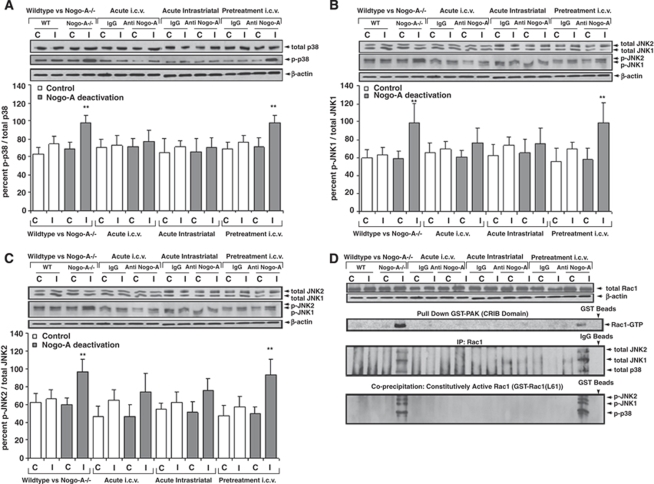
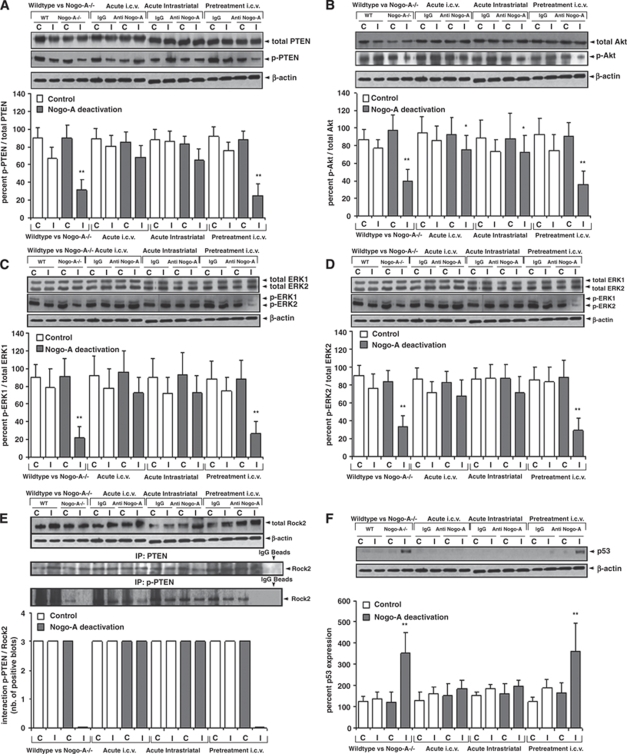
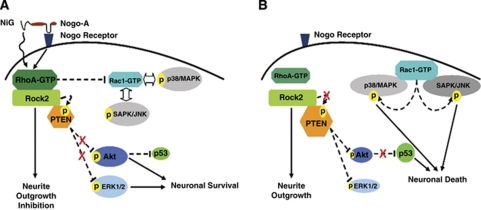
Nogo-A is an oligodendroglial neurite outgrowth inhibitor, the deactivation of which enhances brain plasticity and functional recovery in animal models of stroke. Nogo-A's role in the reperfused brain tissue was still unknown. By using Nogo-A(-/-) mice and mice in which Nogo-A was blocked with a neutralizing antibody (11C7) that was infused into the lateral ventricle or striatum, we show that Nogo-A inhibition goes along with decreased neuronal survival and more protracted neurologic recovery, when deactivation is constitutive or induced 24 h before, but not after focal cerebral ischemia. We show that in the presence of Nogo-A, RhoA is activated and Rac1 and RhoB are deactivated, maintaining stress kinases p38/MAPK, SAPK/JNK1/2 and phosphatase-and-tensin homolog (PTEN) activities low. Nogo-A blockade leads to RhoA deactivation, thus overactivating Rac1 and RhoB, the former of which activates p38/MAPK and SAPK/JNK1/2 via direct interaction. RhoA and its effector Rho-associated coiled-coil protein kinase2 deactivation in turn stimulates PTEN, thus inhibiting Akt and ERK1/2, and initiating p53-dependent cell death. Our data suggest a novel role of Nogo-A in promoting neuronal survival by controlling Rac1/RhoA balance. Clinical trials should be aware of injurious effects of axonal growth-promoting therapies. Thus, Nogo-A antibodies should not be used in the very acute stroke phase.
Figures


Similar articles
-
Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1.J Neurosci. 2002 Dec 1;22(23):10368-76. doi: 10.1523/JNEUROSCI.22-23-10368.2002. J Neurosci. 2002. PMID: 12451136 Free PMC article.
-
BDNF/PI3K/Akt and Nogo-A/RhoA/ROCK signaling pathways contribute to neurorestorative effect of Houshiheisan against cerebral ischemia injury in rats.J Ethnopharmacol. 2016 Dec 24;194:1032-1042. doi: 10.1016/j.jep.2016.11.005. Epub 2016 Nov 8. J Ethnopharmacol. 2016. PMID: 27833029
-
Plasticity-related gene 5 (PRG5) induces filopodia and neurite growth and impedes lysophosphatidic acid- and nogo-A-mediated axonal retraction.Mol Biol Cell. 2010 Feb 15;21(4):521-37. doi: 10.1091/mbc.e09-06-0506. Epub 2009 Dec 23. Mol Biol Cell. 2010. PMID: 20032306 Free PMC article.
-
Targeting the Nogo-A signalling pathway to promote recovery following acute CNS injury.Curr Pharm Des. 2007;13(24):2470-84. doi: 10.2174/138161207781368611. Curr Pharm Des. 2007. PMID: 17692015 Review.
-
The Nogo receptor, its ligands and axonal regeneration in the spinal cord; a review.J Neurocytol. 2002 Feb;31(2):93-120. doi: 10.1023/a:1023941421781. J Neurocytol. 2002. PMID: 12815233 Review.
Cited by
-
Neuronal Nogo-A upregulation does not contribute to ER stress-associated apoptosis but participates in the regenerative response in the axotomized adult retina.Cell Death Differ. 2012 Jul;19(7):1096-108. doi: 10.1038/cdd.2011.191. Epub 2011 Dec 23. Cell Death Differ. 2012. PMID: 22193546 Free PMC article.
-
Encouraging an excitable brain state: mechanisms of brain repair in stroke.Nat Rev Neurosci. 2021 Jan;22(1):38-53. doi: 10.1038/s41583-020-00396-7. Epub 2020 Nov 12. Nat Rev Neurosci. 2021. PMID: 33184469 Free PMC article. Review.
-
Artery reopening is required for the neurorestorative effects of angiotensin modulation after experimental stroke.Exp Transl Stroke Med. 2016 Apr 27;8:4. doi: 10.1186/s13231-016-0018-x. eCollection 2016. Exp Transl Stroke Med. 2016. PMID: 27127602 Free PMC article.
-
MRI Evaluation of Axonal Remodeling After Combination Treatment With Xiaoshuan Enteric-Coated Capsule and Enriched Environment in Rats After Ischemic Stroke.Front Physiol. 2019 Dec 19;10:1528. doi: 10.3389/fphys.2019.01528. eCollection 2019. Front Physiol. 2019. PMID: 31920724 Free PMC article.
-
Persistent increase of Nogo-A-positive cells and dynamic reduction in oligodendroglia lineage cells in white matter regions following experimental and clinical traumatic brain injury.J Neuropathol Exp Neurol. 2025 May 1;84(5):423-435. doi: 10.1093/jnen/nlaf017. J Neuropathol Exp Neurol. 2025. PMID: 40085014 Free PMC article.
References
-
- Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, Tempst P, Strittmatter SM, Sessa WC. A new role for Nogo as a regulator of vascular remodeling. Nat Med. 2004;10:382–388. - PubMed
-
- Bacigaluppi M, Pluchino S, Jametti LP, Kilic E, Kilic U, Salani G, Brambilla E, West MJ, Comi G, Martino G, Hermann DM. Delayed post-ischaemic neuroprotection following systemic neural stem cell transplantation involves multiple mechanisms. Brain. 2009;32:2239–2251. - PubMed
-
- Bergh F, van den Spronk M, Ferreira L, Bloemarts E, Groenink L, Olivier B, Oosting R. Relationship of delay aversion and response inhibition to extinction learning, aggression, and sexual behaviour. Behav Brain Res. 2007;175:75–81. - PubMed
-
- Bregman BS, Kunkel-Bagden E, Schnell L, Dai HN, Gao D, Schwab ME. Recovery from spinal cord injury mediated by antibodies to neurite growth inhibitors. Nature. 1995;378:498–501. - PubMed
-
- Buchli AD, Schwab ME. Inhibition of Nogo: a key strategy to increase regeneration, plasticity and functional recovery of the lesioned central nervous system. Ann Med. 2005;37:556–567. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
