

Genotype-phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I
- PMID: 20087402
- PMCID: PMC2987338
- DOI: 10.1038/ejhg.2009.242
Genotype-phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I
Abstract
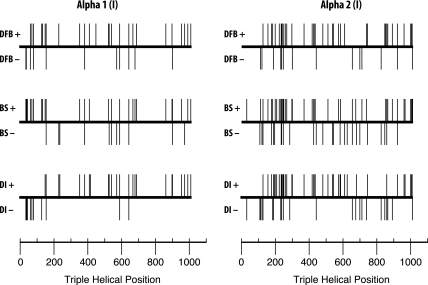
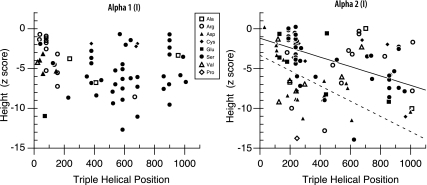
Osteogenesis imperfecta (OI) is a heritable disorder with bone fragility that is often associated with short stature, tooth abnormalities (dentinogenesis imperfecta), and blue sclera. The most common mutations associated with OI result from the substitution for glycine by another amino acid in the triple helical domain of either the alpha1 or the alpha2 chain of collagen type I. In this study, we compared the results of genotype analysis and clinical examination in 161 OI patients (median age: 13 years) who had glycine mutations in the triple helical domain of alpha1(I) (n=67) or alpha2(I) (n=94). Serine substitutions were the most frequently encountered type of mutation in both chains. Compared with patients with serine substitutions in alpha2(I) (n=40), patients with serine substitutions in alpha1(I) (n=42) on average were shorter (median height z-score -6.0 vs -3.4; P=0.005), indicating that alpha1(I) mutations cause a more severe phenotype. Height correlated with the location of the mutation in the alpha2(I) chain but not in the alpha1(I) chain. Patients with mutations affecting the first 120 amino acids at the amino-terminal end of the collagen type I triple helix had blue sclera but did not have dentinogenesis imperfecta. Among patients from different families sharing the same mutation, about 90 and 75% were concordant for dentinogenesis imperfecta and blue sclera, respectively. These data should be useful to predict disease phenotype in newly diagnosed OI patients.
Figures
References
-
- Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
