

An overview of stress response and hypometabolic strategies in Caenorhabditis elegans: conserved and contrasting signals with the mammalian system
- PMID: 20087441
- PMCID: PMC2808051
- DOI: 10.7150/ijbs.6.9
An overview of stress response and hypometabolic strategies in Caenorhabditis elegans: conserved and contrasting signals with the mammalian system
Abstract
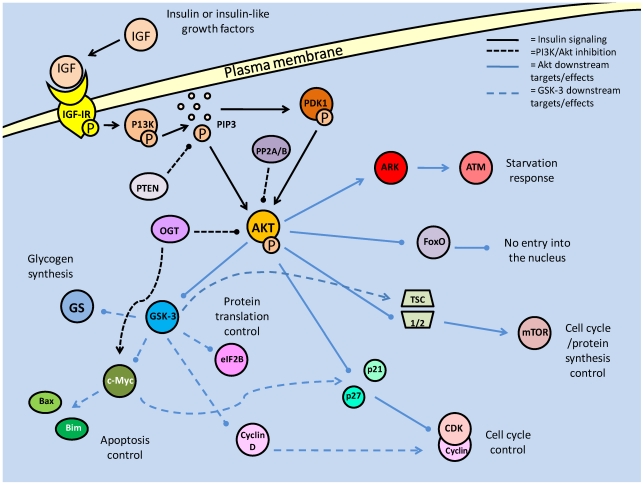
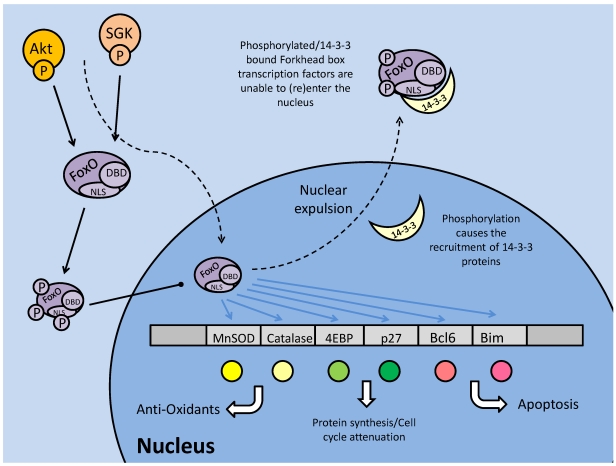
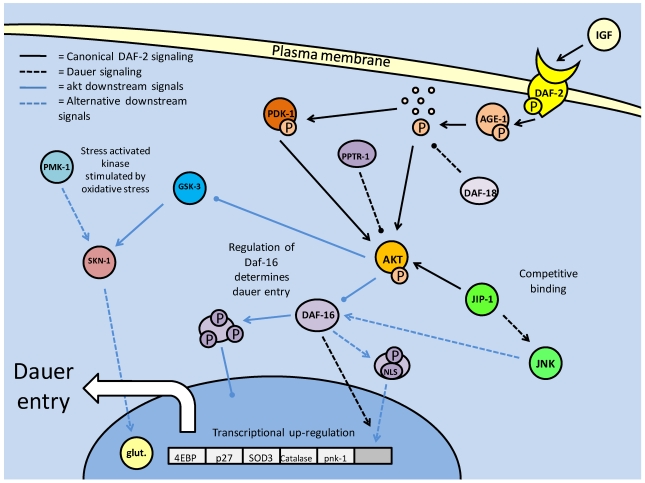
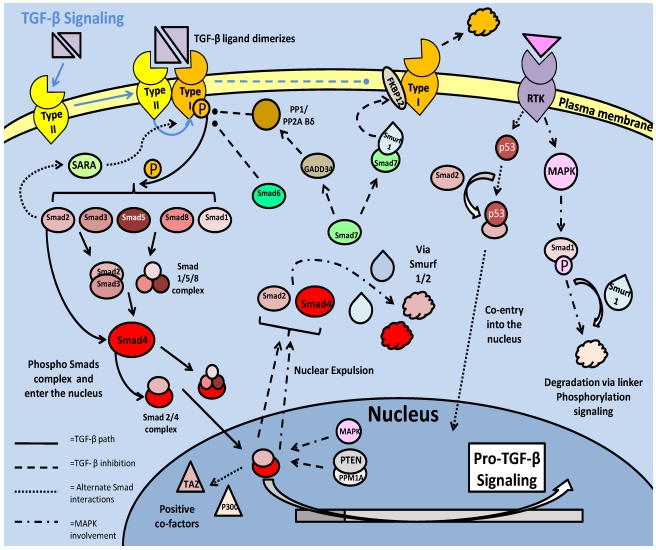
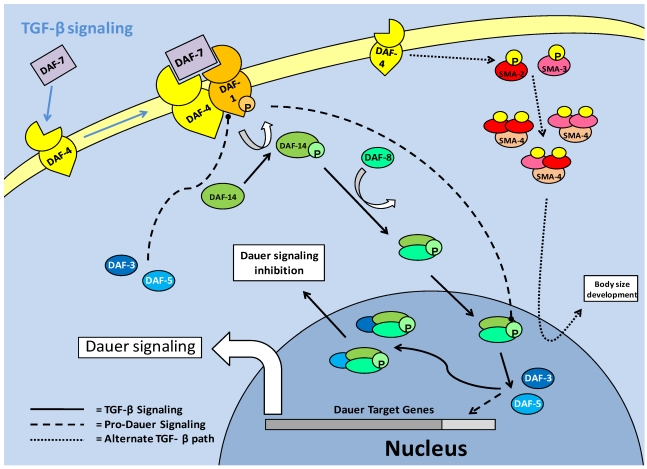
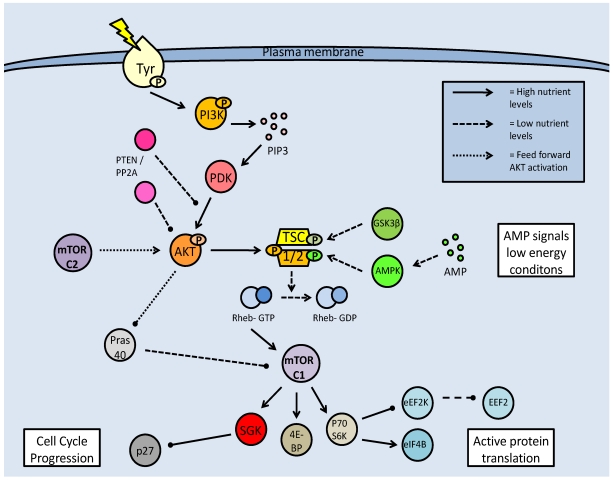
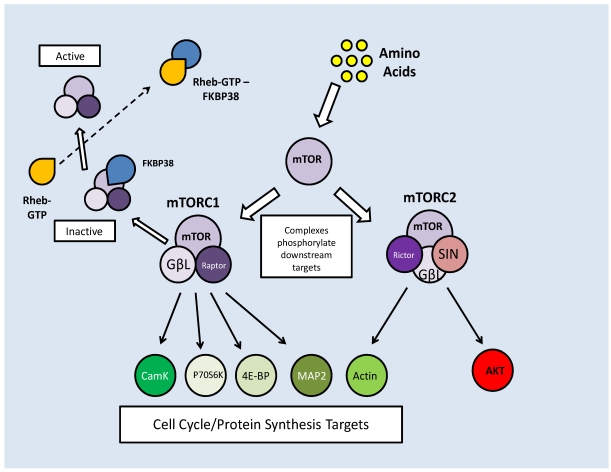
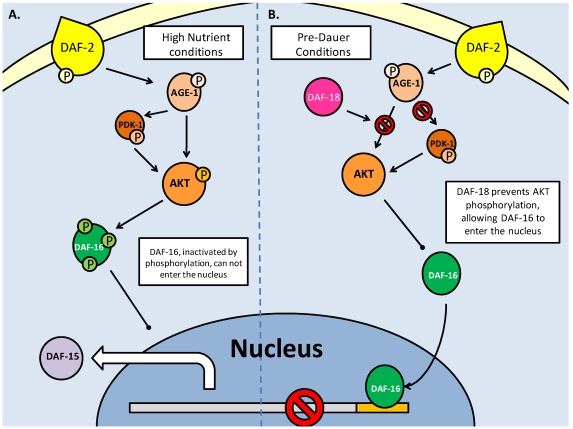
Studies of the molecular mechanisms that are involved in stress responses (environmental or physiological) have long been used to make links to disease states in humans. The nematode model organism, Caenorhabditis elegans, undergoes a state of hypometabolism called the 'dauer' stage. This period of developmental arrest is characterized by a significant reduction in metabolic rate, triggered by ambient temperature increase and restricted oxygen/ nutrients. C. elegans employs a number of signal transduction cascades in order to adapt to these unfavourable conditions and survive for long times with severely reduced energy production. The suppression of cellular metabolism, providing energetic homeostasis, is critical to the survival of nematodes through the dauer period. This transition displays molecular mechanisms that are fundamental to control of hypometabolism across the animal kingdom. In general, mammalian systems are highly inelastic to environmental stresses (such as extreme temperatures and low oxygen), however, there is a great deal of conservation between the signal transduction pathways of nematodes and mammals. Along with conserving many of the protein targets in the stress response, many of the critical regulatory mechanisms are maintained, and often differ only in their level of expression. Hence, the C. elegans model outlines a framework of critical molecular mechanisms that may be employed in the future as therapeutic targets for addressing disease states.
Keywords: Apoptosis; Diapause; Hypometabolism; Lifespan extension; Post-translational modification; Transcriptional regulation.
Conflict of interest statement
Conflict of Interest: The authors have declared that no conflict of interest exists.
Figures
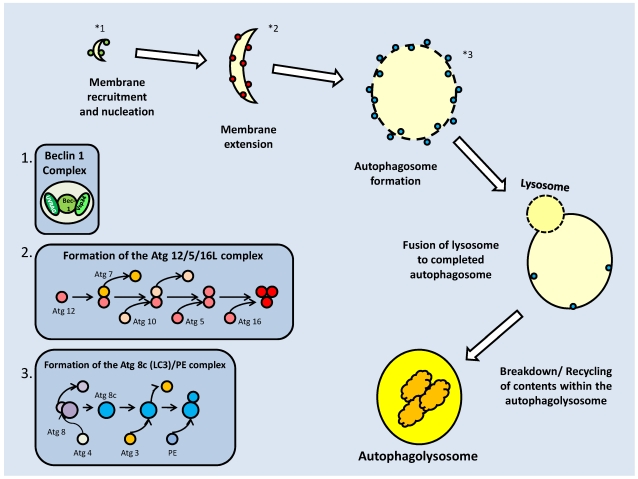
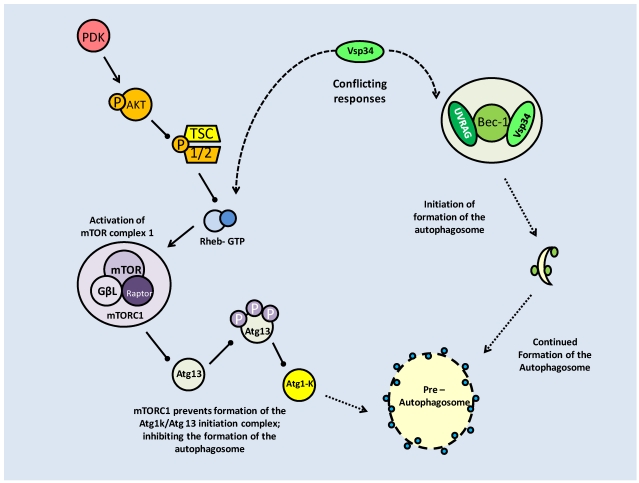
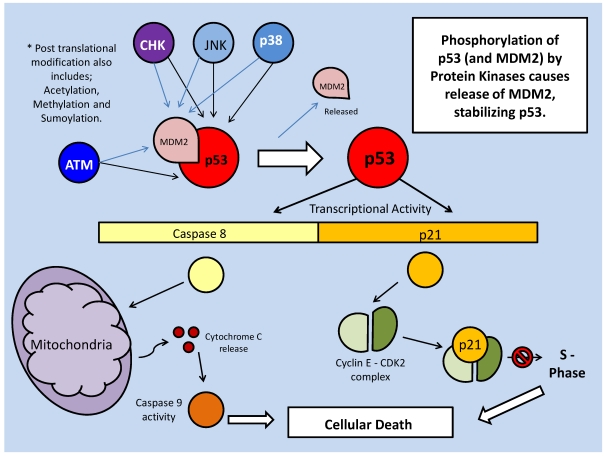
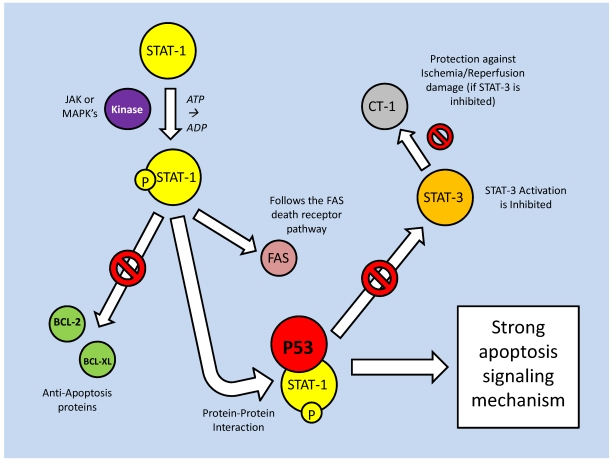
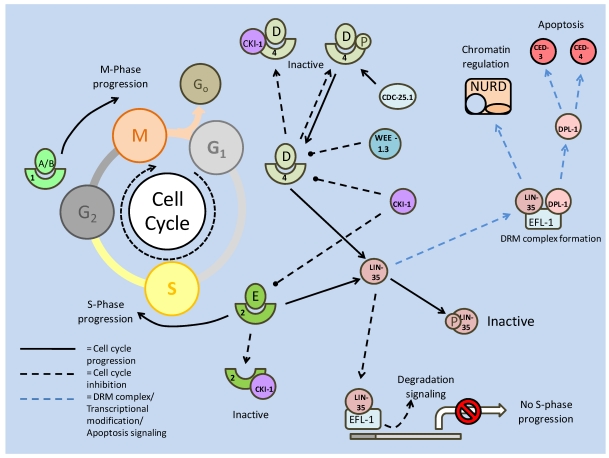
References
-
- Yamashita O, Hasegawa K. Embryonic diapause. In: Kerkut G.A, Gilbert L.I, editors. Comprehensive Insect Physiology, Biochemistry and Pharmacology, vol. 1. Oxford: Pergamon Press; 1985. pp. 407–434.
-
- Nakagaki M, Takei R, Nagashima E, Yaginuma T. Cell cycles in embryos of the silkworm, Bombyx mori: G2-arrest at diapause stage. Roux's Archives of Developmental Biology. 1991;200:223–229. - PubMed
-
- MacRae T.H. Molecular chaperones, stress resistance and development in Artemia franciscana. Semin Cell Dev Biol. 2003;14(5):251–258. - PubMed
-
- Renfree M.B, Shaw G. Diapause. Annu Rev Physiol. 2000;62:353–375. - PubMed
-
- Lopes F.L, Desmarais J.A, Murphy BD. Embryonic diapause and its regulation. Reproduction. 2004;128(6):669–678. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
